| Issue |
Aquat. Living Resour.
Volume 35, 2022
|
|
|---|---|---|
| Article Number | 8 | |
| Number of page(s) | 8 | |
| DOI | https://doi.org/10.1051/alr/2022009 | |
| Published online | 21 June 2022 | |
Research Article
Genetic discrimination of wild versus farmed gilthead sea bream Sparus aurata using microsatellite markers associated with candidate genes
Institute of Oceanography and Fisheries, Split, Croatia
* Corresponding author: This email address is being protected from spambots. You need JavaScript enabled to view it.
Handling Editor: Costas Tsigenopoulos
Received:
16
January
2022
Accepted:
12
April
2022
Abstract
Farm escapees and their offspring impose a significant impact on the environment and may therefore alter the future evolutionary trajectories of wild populations. To date, there is no management plan in place in Mediterranean countries to prevent fish escapes. Here, we investigate microsatellite length variations in three candidate genes, including prolactin (PRL), growth hormone (GH), and the receptor activity modifying protein 3 gene (RAMP3), to study the genetic structure of the main fish species farmed in the Mediterranean, gilthead seabream (Sparus aurata). We also evaluate the performance of microsatellites in discriminating fish origin (wild or farmed). Results from 298 individuals, including farmed, wild adult and juvenile fish were compared with results from 19 neutral markers used in a previous study. All loci were polymorphic, selectively neutral, and had the statistical power to detect significant population differentiation. Global FST was similar to that estimated using 19 loci (0.019 and 0.023, respectively), while pairwise comparisons identified farmed populations as the main drivers of genetic divergence, with a much higher magnitude of overall genetic differentiation within farmed populations (0.076) than that estimated using the 19 neutral microsatellite loci (0.041). Bayesian structural analysis showed that the PRL, GH, and RAMP3 markers were able to distinguish farmed from wild populations, but were not able to distinguish different wild groups as 19 neutral microsatellite markers did. Farmed populations of different origins were assigned to a separate cluster with a high individual assignment score (>88%). It appears that the candidate markers are more influenced by artificial selection compared to neutral markers. Further validation of their efficiency in discriminating wild, farmed, and mixed fish origins using a more robust sample size is needed to ensure their potential use in an escaped fish monitoring programme.
Key words: Gilthead seabream / farm escapes / candidate genes / management
© I. Žužul et al., Published by EDP Sciences 2022
 This is an Open Access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This is an Open Access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
1 Introduction
Environmental sustainability and increased use of marine habitats have been described as the greatest challenges facing the aquaculture industry today (Le Féon et al., 2021). Aquaculture has become the world’s fastest growing food production sectors, though this growth comes with serious concerns for the environment in which it operates. These include pollution from nutrients and chemicals, the spread of diseases, habitat alteration, and interbreeding with wild conspecifics (Buschmann et al., 2006; Ford and Myers, 2008).
Fish escapes from cage farms are common due to construction or operational failures (Jackson et al., 2015). The contribution of escaped fish to natural populations varies widely among farmed species. A recent meta-analysis indicated that one-third of the world’s marine ecoregions are threatened to some degree by escaped farmed fish (Atalah and Sanchez-Jerez, 2020). Namely, escaped fish can have a variety of ecological and genetic impacts on native populations through interbreeding and competition for food and habitat (Jonsson and Jonsson, 2006; Šegvić-Bubić et al., 2011a, 2011b, 2014, 2017). Genetic introgression of escapees can lead to reduced overall fitness of wild populations (Fleming et al., 2000; Glover et al., 2012). Farmed fish typically originate from a small number of parental broodstock characterised by a lower effective population size compared to the wild population. Such a scenario has previously been observed in sea bass and sea bream farming (Brown et al., 2015; Šegvić-Bubić et al., 2017; Žužul et al., 2019; Polovina et al., 2020; Toledo-Guedes et al., 2021). Due to the Ryman-Laikre effect, genetic exchange between captive and wild populations could lead to a reduction in effective population size and genetic diversity of local wild populations (Waples et al., 2016). Depending on the magnitude and frequency of escapes, changes in allele frequency and the introduction of new genetic material may have synergistic effects on certain physiological processes and behaviours of natural populations (Solberg et al., 2020).
Aquaculture of marine fish in Croatia is carried out exclusively in floating cages in coastal or semi-coastal locations. Production of European seabass (Dicentrarchus labrax) and gilthead seabream (Sparus aurata) amounted to 12,000 tonnes in 2019 (FEAP, 2020). Since there is only one national hatchery, most fry are still imported from France and Italy. Unfortunately, many Mediterranean countries, including Croatia, do not have an action plan to prevent and manage escapes (Arechavala-Lopez et al., 2018). Recently, a large number of gilthead sea bream from the eastern Adriatic Sea, consisting of fish of different origins (wild vs. farmed vs. wild farm-associated) and ontogenetic stages (adult vs. juvenile) in 22 populations, were studied using a panel of 19 neutral microsatellite loci in several consecutive years. Hybrids and farmed sea bream were detected at an intermediate level in the natural environment. The total number of escapes was nearly 10%, and more than half were captured in the proximity of aquaculture sites (Žužul et al., 2019). A similar scenario was observed for farmed European seabass from the eastern Adriatic and from the waters of Cyprus, where the proportion of escaped fish in wild samples ranged from 14–15% (Brown et al., 2015; Šegvić-Bubić et al., 2017). This situation indicates the urgent need for a more accurate and cost-effective tool for fishery operators to control farm fish origin and recapture in the case of escape.
Prediction of genes associated with biological traits plays an important role in selection and breeding programmes in aquaculture, where molecular markers have been used to generate linkage maps for important economic phenotypic traits such as growth, sex determination, pathogen resistance, and environmental adaptation (Yue, 2014). Single-nucleotide polymorphisms (SNPs) have become the marker of choice in aquaculture and have proven successful in detecting and managing escaped Atlantic salmon (Karlsson et al., 2016). Microsatellite length variability in candidate genes such as growth hormone (GH) and prolactin (PRL) have the capacity for revealing populations under natural selection (Chaoui et al., 2012). Since these markers have never been tested on gilthead sea bream subjected to artificial selection, the objective of this study was to investigate the potential utility of microsatellites located in candidate genes to identify the origin of farmed fish. Samples were sourced from populations with known genetic backgrounds tested using 19 neutral loci (Žužul et al., 2019) and consisted of farmed, wild adult and juvenile individuals.
2 Materials and methods
2.1 Sampling and microsatellite genotyping
A total of 1586 gilthead seabream were collected in the waters of the eastern Adriatic Sea between July 2015 and January 2017. The dataset included wild juvenile and adult fish, wild farm-associated and adult farmed fish of different broodstock origins (Western and Eastern Adriatic, Ionian Sea and Atlantic Ocean) and farmed in Croatian waters. The farms studied have been working with sourced fingerlings from French, Italian or national hatcheries for more than 20 years and each has an annual production capacity of 100 tonnes. Based on their production, these are important farms in the region. The wild farm-associated fish are samples collected in close proximity to the two largest tuna farms, where they are present year-round and feed on tuna baitfish loss.
Total genomic DNA from fish fins was extracted by proteinase K digestion followed by a simplified DNA isolation procedure (Laird et al., 1991). The total data set was first genotyped with 19 neutral microsatellite markers (SST; Lee-Montero et al., 2013; the results are published in Žužul et al., 2019). For this study, a subset of 298 individuals from nine sites of different origins (Tab. 1, Fig. 1) was genotyped with a multiplex comprising of three microsatellite loci linked to three candidate genes (cgSST), including prolactin (PRL), growth hormone (GH), and receptor activity modifying protein 3 gene (RAMP3) (see Tab. S1; Astola et al., 2003; Launey et al., 2003; Almuly et al., 2005), while genotype data of SST were obtained from the GenoBase of the Institute of Oceanography and Fisheries (http://jadran.izor.hr/~tsegvic/aquapop/GenoBase.html). It should be noted that the current wild population dataset was created with individuals that had with neutral markers in Žužul et al. (2019) a Bayesian assignment score with Structure analysis greater than 90%, representing the fish group of “pure wild origin”. Amplification of cgSST markers was performed in 10 mL reactions, and the final concentrations of all primers were uniformly set at 0.2 μM. PCR conditions were as follows: initial denaturation at 95 °C for 5 min, 26 cycles at 95 °C for 30 s, annealing at 60 °C for 90 s, and elongation at 72 °C for 30 s; and then a final elongation of 30 min at 60 °C. The ABI3130 automated sequencer (Applied Biosystems) was used to separate fragments, while electropherograms and genotypes were analysed using GeneMapper software v.3.5 (Applied Biosystems).
Information of sampling locations, year and population code, along with the origin and number of individual gilthead seabream that were genetically assayed with 3 microsatellites linked to three candidate genes (cgSST).
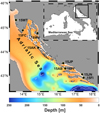 |
Fig. 1 Sampling locations of wild adult gilthead seabream (WT, Tar Bay; WV, Velebit Channel), wild juveniles (JP, Pantana River; JN, Neretva River), farm-associated adults (AK, Mrdina; AB, Maslinova) and farmed adults (15FC, Budava Bay; 15FI, Mali Ston Bay; 16FF Maslinova Bay) |
2.2 Genetic diversity and population structuring
The presence and frequency of null alleles were tested using MICROCHECKER 2.2.3 software (Van Oosterhout et al., 2004) and FREENA (Chapuis and Estoup, 2007), respectively. GENEPOP v.3.1b (Raymond and Rousset, 2003) was used to perform an exact test for linkage disequilibrium (LD) and to test for significant deviations from Hardy-Weinberg equilibrium (HWE) for each locus and population. The Bonferroni correction (Rice, 1989) was applied to adjust significance levels for multiple tests. The mean effective number of alleles across all loci (Ae) and the mean number of alleles per locus (A) were calculated using POPGENE v.1.32 (Yeh et al., 2000). The inbreeding coefficient (FIS) and allelic richness (Ar) were calculated using FSTAT v.2.3 (Goudet et al., 2002), while the polymorphism information content (PIC) of each locus was estimated using Cervus v3.0.3 (Kalinowski et al., 2007). ARLEQUIN v.3.5 software was used to calculate observed (Ho) and expected (He) heterozygosity. Deviation from neutrality was performed in LOSITAN (Antao et al., 2008) using the FST method for outlier detection (Beaumont and Nichols, 1996) for candidate loci. A stepwise mutation model was applied with the following settings: 50,000 simulations, 95% CI, forced mean FST with a false discovery rate of 0.05.
Statistical power of genetic homogeneity tests for the set of markers and sample size was evaluated using POWSIM (Ryman et al., 2006). Pairwise and global FST values were calculated using ARLEQUIN v.3.5, with statistical significance assessed after 10,000 permutations. The Bayesian clustering program STRUCTURE 2.3.4 (Pritchard et al., 2000) was used to analyse the genetic structure of the included populations, with the parameters: population admixture and a priori location (LOCPRIOR) with a burn-in of 10,000 interactions followed by a run of 500,000 MCMC steps and K values from 1 to the maximum number of groups examined, with 20 replicates each. The most likely number of clusters was determined using the ad?hoc statistic deltaK implemented in Structure Harvester 0.6.93 (Earl and von Holdt, 2012) and visualised using CLUMPP (Jakobsson and Rosenberg, 2007) and DISTRUCT (Rosenberg, 2004). The same analysis was repeated for i) the data set of 19 SST genotypes, and ii) the dataset combining the genotypes of the three cgSSTs and 19 SSTs for 298 individuals each. In addition, a non-model-based discriminant analysis of principal components (DAPC) method implemented in the Adegenet package (Jombart, 2008) for R was applied to the original and combined datasets, with the retained number of principal components (PCs) optimised using the xvalDapc function (Jombart, 2008).
3 Results
3.1 Genetic diversity
A total of 298 Sparus aurata individuals were genotyped with three cgSST microsatellite markers, while genotype data for the SST dataset were obtained from GenoBase, IOR (Tab. 1, Fig. 1). The percentage of missing data per locus ranged from 0 to 1.5%, with an average of 0.3% and 0.8% for the SST and cgSST datasets, respectively.
Several populations showed significant deviation from Hardy-Weinberg equilibrium with trends toward heterozygote deficiency at the candidate gene locus (PRL) (Tab. S2, Supplementary information). The PRL locus showed the presence of null alleles at a low frequency (<8%), and this locus was retained because the estimated FST values with and without application of the ENA correction method were comparable; 0.019 vs. 0.018 for FST with vs. without ENA, respectively, with overlapping 95% CI. For the neutral loci (SST), the estimated FST values with and without application of the ENA correction method were also comparable (0.0292 vs. 0.0291, respectively), with overlapping 95% CI.
All loci examined were polymorphic (mean PIC > 0.5), with the number of alleles per locus ranging from 4 to 38 for the SST and from 10 to 21 for the cgSST dataset (Tab. S2). The expected heterozygosity (He) ranged from 0.73 to 0.81 for the SST dataset and from 0.65 to 0.84 for He of the cgSST dataset. The observed heterozygosity (Ho) was similar among the studied populations (0.72–0.73) for the cgSST dataset, whereas Ho ranged from 0.76 to 0.72 for the SST dataset, with the lowest value found in the farmed group (Tab. 2). Concerning the cgSST loci examined, only the effective number of alleles per locus (5.1 vs. 8.0) and allelic richness (10.1 vs. 7.5) were found to be significantly reduced in the farmed populations in contrast to wild populations. The inbreeding coefficient, FIS, ranged from 0.01 to 0.08 in the SST dataset and was significantly greater than zero in 2 out of the 9 populations in the cgSST dataset, ranging from –0.03 to 0.12 (Tab. 2). Among the cgSST loci, the PRL locus showed a different distribution of alleles when populations were grouped by origin (wild vs. farmed), where alleles 328 and 344 were more frequent in the farmed than in the wild group (22 vs. 9%; 23 vs. 7%, respectively). No major changes in allele frequency distribution between fish origins were detected for the other two loci (Fig. S1).
Summary statistic for genetic variation of gilthead seabream Sparus aurata in the Adriatic Sea showing the average number of alleles (A), effective number of alleles (Ae), allelic richness (Ar), expected (He) and observed (Ho) heterozygosity, and fixation index (FIS), for 19 neutral SST loci and 3 candidate gene loci (cgSST).
3.2 Genetic differentiation among populations
The performance of cgSST markers to detect significant population differences was high. Simulations in POWSIM yielded a 95% probability (χ2, Fisher’s test) of detecting a true differentiation of FST = 0.005 under different scenarios of effective population size (Ne) and number of drift generations (t) with 1000 replications (Tab. S3). Following tests of neutrality using the LOSITAN program, all three loci linked to candidate genes fell into the neutral category. The global FST across all nine populations was 0.023 (p < 0.001) for the SST dataset and 0.019 (p < 0.001) for the cgSST dataset, demonstrating a low to moderate level of differentiation between populations. Pairwise FST across all gilthead seabream samples ranged from –0.003 to 0.085 (Tab. 3), with 29 of 36 pairwise comparisons significant for the SST dataset and 18 of 36 significant for the cgSST dataset (p < 0.001 after Bonferroni correction). All significant cgSST comparisons were found between the farmed and the other populations, as well as among the three farmed populations, while an additional significant comparison was found between wild adults and wild farm-associated and juveniles for the SST dataset (Tab. 3).
Structure analysis of the three cgSST dataset of nine gilthead seabream populations revealed the presence of four clusters (K = 4, Fig. 2a). All farmed populations of different origins (CRO, ITA, FR) were each assigned to a separate cluster (blue, yellow, and red) with a high individual assignment score (≥88%) for each cluster. In contrast, wild populations from different sources (adults, juveniles, farm-associated) showed homogeneity and were assigned to the fourth cluster (green) with high assignment values ranging from 0.6 to 0.95% (Tab. S4). When hierarchical structuring was applied, no further structure was detected within the wild cluster. Structure analysis of the dataset containing 19 SSTs and the dataset in which the genotypes of 19 SSTs were added to those of the three cgSSTs revealed K = 5 (Fig. 2b, Fig. S2). This corresponds to the same pattern for the farm populations as for the cgSST dataset, though the wild populations that previously showed homogeneity in the cgSST dataset were assigned to two separate clusters (orange and green; Fig. 2b). The orange cluster included only wild-sampled populations, while the green cluster included wild farm-associated populations and juveniles, respectively. The DAPC analysis showed a similar pattern of clustering to that observed in STRUCTURE (Fig. S3).
Pairwise FST values based on 3 cgSST loci (below diagonal) and pairwise FST values based on 19 neutral SST loci (above diagonal) among 9 populations of gilthead seabream from the Adriatic Sea, including wild, farmed, farm-associated and wild juvenile populations. Significant FST values are underlined at p < 0.001 (Bonferroni correction). Population codes are explained in Table 1.
 |
Fig. 2 Bayesian STRUCTURE clustering of the gilthead seabream reduced dataset based on (a) 3 cgSST loci and (b) combined 3 cgSST and 19 neutral SST loci, assuming four (K = 4) and five (K = 5) inferred clusters. Vertical coloured lines represent individual admixture coefficients. Black horizontal lines divide individuals from different sampling sites. See Tab. 1 for abbreviated sample names. |
4 Discussion
In the present study, three cgSST microsatellite markers were used for origin screening of 298 specimens of sea bream (wild adults and juveniles, farmed associated adults, and farmed individuals of different broodstock origin) collected in the coastal region of the eastern Adriatic Sea. Data obtained on the same individuals were compared with the results of 19 putative neutral markers (Žužul et al., 2019). These cgSST markers were selected because (1) their molecular functions are associated with traits of high interest for aquaculture (growth rate, osmoregulatory capacity) and are therefore subject to artificial selection in farmed fish, and (2) the GH and PRL genes have known effects on quantitative traits, e.g., growth rate or osmoregulatory capacity in fish, where several authors have linked the length variation of a polymorphic microsatellite in the promoter region of a gene to its transcriptional activity (Almuly et al., 2005, 2008; Astola et al., 2003; Blel et al., 2010; He et al., 2012).
Candidate gene markers were identified as selectively neutral in the present study, in contrast to previous reports on GH and PRL loci when habitat-based genetic differentiation was tested in gilthead seabream (Chaoui et al., 2012; Guinand et al., 2016), but their power to detect significant population differentiation between wild and farmed populations was high. Indeed, farmed populations were the main driver of genetic divergence (Tab. 3), as observed in previous studies (Šegvić-Bubić et al., 2011b; Loukovitis et al., 2012; Cossu et al., 2019), although the magnitude of overall genetic differentiation within farmed populations was much larger (0.076) than that estimated with 19 neutral microsatellite loci (0.041; Žužul et al., 2019). For both types of markers, significantly lower allelic richness, effective size, and heterozygosity were observed in farmed populations compared with wild populations (Tab. 2). Such a pattern of indices has been reported for several fish species (Glover et al., 2013; Šegvić-Bubić et al., 2017; Cossu et al., 2019) and generally reflects high variance in the family size of breeding populations used in hatcheries and, in the case of gilthead seabream, a bias in the contribution of males and females to each mass spawning event (Brown, 2003). Reduced effective population size due to limited broodstock size may favour both the occurrence of the founder effect and genetic drift, which in turn may lead to a loss of genetic diversity (Loukovitis et al., 2012) and consequently limit the potential for selective breeding. The extent to which diversity is lost depends on several factors, including the number, size, and origin of broodstocks and spawning methods (García-Fernández et al., 2018).
While the polymorphism information content (PIC) for the PRL locus was the highest among the cgSST loci, only PRL showed significantly positive FIS values exclusively affecting wild populations (Tabs. S1 and S2). The presence of heterozygote deficiencies, which can be partially explained by the presence of null alleles, was previously noted for the candidate gene when genetic variation patterns in relation to habitat type were studied in wild gilthead sea bream (Chaoui et al., 2012). Furthermore, only the PRL locus showed a bias in the distribution of allele frequencies between the pooled wild and farmed populations, and the contribution of two alleles (328 and 344) was significantly increased in the farmed populations in contrast to wild populations (Fig. S1). The discrepancy in the distribution of allele frequencies within wild populations represented by adults and juveniles from different areas of the eastern Adriatic was not captured.
With regard to the genetic structuring of the populations tested in this study, Bayesian structural analysis showed that the markers PRL, GH, and RAMP3 were able to distinguish farmed from wild populations and, in addition, to assign farmed populations of different origins (CRO, ITA, FR) to a separate cluster with a high individual assignment score (>88%, Fig. 2, Tab. S4). However, the tested markers were not able to distinguish different wild groups (wild adults, juveniles and farm-associated) identified when using 19 neutral microsatellite markers (Fig. S2). It appears that candidate markers are more affected by artificial selection and therefore have great potential to be used as a tool for farm origin screening in an escaped fish surveillance programme. The lack of genetic structure within wild adults (15WT and 16WV) and reduced genetic connectivity with wild offspring and tuna farm-associated collections was also reported by Žužul et al. (2019) with a larger dataset. It could be argued that wild populations sampled over several years from large shoals that first emerge in the northern Adriatic in autumn and then migrate to the southern parts, perform exclusively trophic north-south migrations along the eastern Adriatic coast, and spawning occurs elsewhere in the Mediterranean. On the other hand, Žužul et al. (2019) confirmed a perennial genetic connection of wild fish associated with tuna farms and offspring from the coastal nursery areas, suggesting that tuna farm environments act as spawning grounds for resident sea bream. The permanent and very frequent aggregation behaviour of sea bream in the bottom layers of semi-offshore tuna farms (Stagličić et al., 2017) was primarily attributed to the FAD effect and the increased availability of tuna baitfish losses composed of small pelagic species.
It should be noted that cases of farm escapees or hybrids in the wild fish origin observed with 19 neutral markers (Žužul et al., 2019) were intentionally omitted from the present study. The wild population dataset was created with individuals of pure origin, i.e., individuals that had a Bayesian assignment score with neutral markers greater than 90%, because the goal was to first test the performance of the three loci in assigning individuals to their origin. Such a design limited the ability to evaluate the performance of candidate genes to the detection of wild and farmed hybrids and introgression per se. As the assignments of farm individuals were consistent with the results of 19 putative neutral markers, further validation should be performed on individuals of mixed origin. This step is very important for the management of escaped fish to clarify whether the candidate markers reflect the admixture pattern of the putative neutral loci, which in turn allows for the detection of hybrids.
Complementary to the genetic approach, a variety of origin descriptors are available to identify escaped gilthead seabream in wild populations. The simplest yet cheapest way to distinguish fish origin is to use external characteristics in combination with morphometrics (e.g., body and otolith shape, condition index) and organoleptic descriptors, but their use is limited to early escaped fish (Arechavala-Lopez et al., 2013; Šegvić-Bubić et al., 2014; Talijančić et al., 2019, 2021). Recent studies have shown that when analysing body shape and otolith characteristics, attribution of farm seabream to their true origin is more than 75% correct (Talijančić et al., 2019, 2021), while Šegvić-Bubić et al. (2020) showed that the pattern of scale shape, supported by scale microchemistry, can serve as a discriminator in controlling recent escapes.
The application of diagnostic SNP arrays is routinely used in the Norwegian monitoring programme to investigate the extent of farmed escapees and introgression into wild salmon populations (Karlsson et al., 2011, 2014, 2016; Diserud et al., 2020). The pace of genetic progress in species important for Mediterranean aquaculture has recently accelerated through the work of several European projects. The recently developed and publicly available tools for high-throughput genotyping of ~30 K SNPs for European seabass and gilthead seabream (Peñaloza et al., 2021) as well as those in Griot et al. (2021) provide a new tool that can be used to assess the genetic contribution of aquaculture escapees to wild populations. Surprisingly, none of the regulatory frameworks for aquaculture in the Mediterranean includes the management of escaped fish, in contrast to countries with highly developed marine aquaculture such as Norway, Canada or Chile.
In the present study, selected cgSST markers demonstrated their potential to discriminate the origin of fish (wild vs. farmed, Bayesian score assignment >88%), although selective breeding of gilthead seabream is still in its infancy and strains of several generations are under mass or family selection (Janssen et al., 2017). Nevertheless, further validation of their efficiency using a larger sample is needed to ensure their usefulness for the management of escaped gilthead sea bream.
Altogether, there is a solid knowledge base that can become an integral part of the management system for escaped fish in the Mediterranean Sea. In coastal areas, the spatial distribution of escaped fish is closely associated with fish farms (Šegvić-Bubić et al., 2017), where fish remain near the farm for several weeks to adapt. This time frame is considered the most appropriate for recapture of escaped fish, as described in Izquierdo-Gomez et al. (2016), where almost half of the biomass of escaped fish was captured in the first days after escape. Therefore, identification and recapture of escaped fish is an important step to address this emerging problem.
Supplementary Material
Acknowledgements
This study was fully supported by the Croatian Science Foundation under project IP-2014-09-9050. The authors are grateful to local recreational fisherman for assistance with sample collection.
References
- Almuly R, Poleg-Danin Y, Gorshkov S, Gorshkova G, Rapoport B, Soller M, Funkenstein B. 2005. Characterization of the 5′ flanking region of the growth hormone gene of the marine teleost, gilthead sea bream Sparus aurata: analysis of a polymorphic microsatellite in the proximal promoter. Fish Sci 71: 479–490. [CrossRef] [Google Scholar]
- Almuly R, Skopal T, Funkenstein B. 2008. Regulatory regions in the promoter and first intron of Sparus aurata growth hormone gene: repression of gene activity by a polymorphic minisatellite. Comp Biochem Physiol Part D Genomics 3: 43–50. [Google Scholar]
- Antao T., Lopes A., Lopes R.J., Beja-Pereira A., Luikart G. 2008. LOSITAN: a workbench to detect molecular adaptation based on a Fst-outlier method. BMC Bioinform 9: 1–5. [CrossRef] [Google Scholar]
- Arechavala-Lopez P., Fernandez-Jover D., Black K.D., Ladoukakis E., Bayle-Sempere J.T., Sanchez-Jerez P., Dempster T. 2013. Differentiating the wild or farmed origin of Mediterranean fish: a review of tools for sea bream and sea bass. Rev Aqua 4: 1–21. [Google Scholar]
- Arechavala-Lopez P., Toledo-Guedes K., Izquierdo-Gomez D., Šegvić-Bubić T., Sanchez-Jerez P. 2018. Implications of sea bream and sea bass escapes for sustainable aquaculture management: a review of interactions, risks and consequences. Rev Fish Sci Aquac 26: 214–234. [CrossRef] [Google Scholar]
- Astola A., Orti, M., Calduch-Giner J.A., Pérez-Sánchez J., Valdivia M.M. 2003. Isolation of Sparus auratus prolactin gene and activity of the cis-acting regulatory elements. Gen Comp Endocrinol 134: 57–61. [CrossRef] [PubMed] [Google Scholar]
- Atalah J., Sanchez-Jerez P. 2020. Global assessment of ecological risks associated with farmed fish escapes. Glob Ecol Conserv DOI: 10.1016/j.gecco.2019.e00842. [Google Scholar]
- Beaumont M.A., Nichols R.A. 1996. Evaluating loci for use in the genetic analysis of population structure. Proc Royal Soc B 263: 1619–1626. [CrossRef] [Google Scholar]
- Blel H., Panfili J., Guinand B., Berrebic P., Saida K., Durandb J.D. 2010. Selection footprint at the first intron of the Prl gene in natural populations of the flathead mullet (Mugil cephalus, L. 1758). J Exp Mar Biol Ecol 387: 60–67. [CrossRef] [Google Scholar]
- Brown R.C. 2003. Genetic management and selective breeding in farmed populations of gilthead seabream, Sparus aurata. PhD thesis, University of Stirling, U.K. [Google Scholar]
- Brown C., Miltiadou D., Tsigenopoulos C.S. 2015. Prevalence and survival of escaped European seabass Dicentrarchus labrax in Cyprus identified using genetic markers. Aquac Environ Interact 7: 49–59. [CrossRef] [MathSciNet] [Google Scholar]
- Buschmann A.H., Riquelme V.A., Hernández-González M.C., Varela D., Jiménez J.E., Henríquez L.A., Vergara P.A., Guíñez R., Filún L. 2006. A review of the impacts of salmonid farming on marine coastal ecosystems in the southeast Pacific. ICES J Mar Sci 63: 1338–1345. [CrossRef] [Google Scholar]
- Chaoui L., Gagnaire P.A., Guinand B., Quignard J.P., Tsigenopoulos C., Kara M.H., Bonhomme F. 2012. Microsatellite length variation in candidate genes correlates with habitat in the gilthead sea bream Sparus Aurata. Mol Ecol 21: 5497–5511. [CrossRef] [PubMed] [Google Scholar]
- Chapuis M.P., Estoup A. 2007. Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol 24: 621–631. [CrossRef] [PubMed] [Google Scholar]
- Cossu P., Scarpa F., Sanna D., Lai T., Dedola G.L., Curini-Galletti M., Mura L., Fois N., Casu M. 2019. Influence of genetic drift on patterns of genetic variation: the footprint of aquaculture practices in Sparus aurata (Teleostei: Sparidae). Mol Ecol 28: 3012–3024. [CrossRef] [PubMed] [Google Scholar]
- Diserud O.H., Hedger R., Finstad B., Hendrichsen D., Jensen A.J., Ugedal O. 2020. Salmon louse infestation in wild brown trout populations generates multi-modal mixture distributions. Aquac Environ Interact 12: 447–456. [CrossRef] [Google Scholar]
- Earl D.A., von Holdt B.M. 2012. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4: 359–361. [CrossRef] [Google Scholar]
- FEAP. 2020. Federation of European Aquaculture Producers, annual report. [Google Scholar]
- Fleming I.A., Hindar K., Mjölneröd I.B., Jonsson B., Balstad T., Lamberg A. 2000. Lifetime success and interactions of farm salmon invading a native population. Proc Royal Soc B 267: 1517–1523. [CrossRef] [PubMed] [Google Scholar]
- Ford J.S., Myers R.A. 2008. A global assessment of salmon aquaculture impacts on wild salmonids. PLoS Biol DOI: 10.1371/journal.pbio.0060033. [PubMed] [Google Scholar]
- García-Fernández C., Sánchez J.A., Blanco G. 2018. Early assessment of gilthead sea bream (Sparus aurata) spawning dynamics by mini-broodstocks. Aquac Res 49: 36–47. [CrossRef] [Google Scholar]
- Glover K.A., Quintela M., Wennevik V., Besnier F., Sørvik A.G., Skaala Ø. 2012. Three decades of farmed escapees in the wild: a spatio-temporal analysis of Atlantic salmon population genetic structure throughout Norway. PLoS One DOI: 10.1371/journal.pone.0043129. [Google Scholar]
- Glover K.A., Pertoldi C., Besnier F., Wennevik V., Kent M., Skaala Ø. 2013. Atlantic salmon populations invaded by farmed escapees: quantifying genetic introgression with a Bayesian approach and SNPs. BMC Genet 14: 1–19. [PubMed] [Google Scholar]
- Goudet J. 2002. FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9.3.2) https://www2.unil.ch/popgen/softwares/fstat.htm [Google Scholar]
- Griot R., Allal F., Phocas F., Brard-Fudulea S., Morvezen R., Haffray P., François Y., Morin T., Bestin A., Bruan, J.-S., Cariou S., Peyrou B., Brunier J., Vandeputte M. 2021. Optimization of genomic selection to improve disease resistance in two marine fishes, the European Sea Bass (Dicentrarchus labrax) and the Gilthead Sea Bream (Sparus aurata). Front Genet 12: 1294. [Google Scholar]
- Guinand B., Chauvel C., Lechene M., Tournois J., Tsigenopoulos C.S., Darnaude A.M., Gagnaire P.A. 2016. Candidate gene variation in gilthead sea bream reveals complex spatiotemporal selection patterns between marine and lagoon habitats. Mar Ecol Prog Ser 558: 115–127. [CrossRef] [Google Scholar]
- He X.P., Xia J.H., Wang C.M., Pang H.Y., Yue G.H. 2012. Significant associations of polymorphisms in the prolactin gene with growth traits in Asian seabass (Lates calcarifer). Anim Genet 43: 233–236. [CrossRef] [PubMed] [Google Scholar]
- Izquierdo-Gomez D., Sanchez-Jerez P. 2016. Management of fish escapes from Mediterranean Sea cage aquaculture through artisanal fisheries. Ocean Coast Manag 122: 57–63. [CrossRef] [Google Scholar]
- Jackson D., Drumm A., McEvoy S., Jensen Ø., Mendiola D., Gabiña G., Borg J.A., Papageorgiou N., Karakassis Y., Black K.D. 2015. A pan-European valuation of the extent, causes and cost of escape events from sea cage fish farming. Aquaculture 436: 21–26. [CrossRef] [Google Scholar]
- Jakobsson M., Rosenberg N.A. 2007. CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23: 1801–1806. [CrossRef] [PubMed] [Google Scholar]
- Janssen K., Chavanne H., Berentsen P., Komen H. 2017. Impact of selective breeding on European aquaculture. Aquaculture 472: 8–16. [CrossRef] [Google Scholar]
- Jombart T. 2008. Adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24: 1403–1405. [CrossRef] [PubMed] [Google Scholar]
- Jonsson B., Jonsson N. 2006. Cultured Atlantic salmon in nature: a review of their ecology and interaction with wild fish. ICES J Mar Sci 63: 1162–1181. [CrossRef] [Google Scholar]
- Kalinowski S.T., Taper M.L., Marshall T.C. 2007. Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment. Mol Ecol 16: 1099–1106. [CrossRef] [PubMed] [Google Scholar]
- Karlsson S., Moen T., Lien S., Glover K.A., Hindar K. 2011. Generic genetic differences between farmed and wild Atlantic salmon identified from a 7K SNP‐chip. Mol Ecol Resour 11: 247–253. [CrossRef] [PubMed] [Google Scholar]
- Karlsson S., Diserud O.H., Moen T., Hindar K. 2014. A. standardized method for quantifying unidirectional genetic introgression. Ecol Evol 4: 3256–3263. [CrossRef] [PubMed] [Google Scholar]
- Karlsson S., Diserud O.H., Fiske P., Hindar K. 2016. Widespread genetic introgression of escaped farmed Atlantic salmon in wild salmon populations. ICES J Mar Sci 73: 2488–2498. [CrossRef] [Google Scholar]
- Laird P.W., Zijderveld A., Linders K., Rudnicki M.A., Jaenisch R., Berns A. 1991. Simplified mammalian DNA isolation procedure. Nucleic Acids Res 19: 4293. [CrossRef] [PubMed] [Google Scholar]
- Launey S., Krieg F., Haffray P. 2003. Twelve new microsatellite markers for gilthead seabream (Sparus aurata L.): characterization, polymorphism and linkage. Mar Environ Res 3: 457–459. [Google Scholar]
- Le Féon S., Dubois T., Jaeger C., Wilfart A., Akkal-Corfini N., Bacenetti J., Costantini M., Aubin J. 2021. DEXiAqua, a model to assess the sustainability of aquaculture systems: methodological development and application to a French Salmon Farm. Sustainability DOI: 10.3390/su13147779. [Google Scholar]
- Lee-Montero I., Navarro A., Borrell Y., García-Celdrán M., Martín N., Negrín-Báez D., Blanco G., Armero E., Berbel C., Zamorano M.J., Sánchez J.J., Estévez A., Ramis G., Manchado M., Afonso J.M. 2013. Development of the first standardised panel of two new microsatellite multiplex PCRs for gilthead seabream (Sparus aurata L.). Anim Genet 44: 533–546. [CrossRef] [PubMed] [Google Scholar]
- Loukovitis D., Sarropoulou E., Vogiatzi E., Tsigenopoulos C.S., Kotoulas G., Magoulas A., Chatziplis D. 2012. Genetic variation in farmed populations of the gilthead sea bream Sparus aurata in Greece using microsatellite DNA markers. Aquac Res 43: 239–246. [CrossRef] [Google Scholar]
- Peñaloza C., Manousaki T., Franch R., Tsakogiannis A., Sonesson A.K., Aslam M.L., Allal F., Bargelloni L., Houston R.D., Tsigenopoulos C.S. 2021. Development and testing of a combined species SNP array for the European seabass (Dicentrarchus labrax) and gilthead seabream (Sparus aurata). Genomics 113: 2096–2107. [CrossRef] [PubMed] [Google Scholar]
- Polovina E.-S., Kourkouni E., Tsigenopoulos C.S., Sanchez-Jerez P., Ladoukakis E.D. 2020. Genetic structuring in farmed and wild Gilthead seabream and European seabass in the Mediterranean Sea: implementations for detection of escapees. Aquat. Living Resour. 33: 7 [Google Scholar]
- Pritchard J.K., Stephens M., Donnelly P. 2000. Inference of population structure using multilocus genotype data. Genetics 155: 945–959. [CrossRef] [PubMed] [Google Scholar]
- Raymond M., Rousset F. 2003. A population genetic software for exact test and ecumenicism: GENEPOP, version 3.4. Heredity 68: 248–249. [Google Scholar]
- Rice W.R. 1989. Analyzing tables of statistical tests. Evolution (N. Y) 43: 223–225. [Google Scholar]
- Rosenberg N.A., Calabrese P.P. 2004. Polyploid and multilocus extensions of the Wahlund inequality. Theor Popul Biol 66: 381–391. [CrossRef] [PubMed] [Google Scholar]
- Ryman N., Palm S. 2006. POWSIM: a computer program for assessing statistical power when testing for genetic differentiation. Mol Ecol Notes 6: 600–602. [CrossRef] [Google Scholar]
- Solberg M.F., Robertsen G., Sundt-Hansen L.E., Hindar K., Glover K.A. 2020. Domestication leads to increased predation susceptibility. Sci Rep 10: 1–11. [CrossRef] [PubMed] [Google Scholar]
- Šegvić-Bubić T., Grubišić L., Karaman N., Tičina V., Mišlov Jelavić K., Katavić I. 2011a. Damages on mussel farms potentially caused by fish predation-Self service on the ropes? Aquaculture 319: 497–504. [CrossRef] [Google Scholar]
- Šegvić-Bubić T., Lepen I., Trumbić Ž., Ljubković J., Sutlović D., Matić-Skoko S., Grubišić L., Glamuzina B., Mladineo I. 2011b. Population genetic structure of reared and wild gilthead sea bream (Sparus aurata) in the Adriatic Sea inferred with microsatellite loci. Aquaculture 318: 309–315. [CrossRef] [Google Scholar]
- Šegvić-Bubić T., Talijančić I., Grubišić L., Izquierdo-Gomez D., Katavić I. 2014. Morphological and molecular differentiation of wild and farmed gilthead sea bream Sparus aurata: implications for management. Aquac Environ Interact 6: 43–54. [CrossRef] [Google Scholar]
- Šegvić-Bubić T., Grubišić L., Trumbić Ž., Stanić R., Ljubković J., Maršić-Lučić J., Katavić I. 2017. Genetic characterization of wild and farmed European seabass in the Adriatic Sea: assessment of farmed escapees using a Bayesian approach. ICES J Mar Sci 74: 369–378. [CrossRef] [Google Scholar]
- Šegvić-Bubić T., Talijančić I., Vulić L., Šegvić B., Žužul I., Radonić I., Grubišić L. 2020. Assignment of gilthead seabream Sparus aurata to its origin through scale shape and microchemistry composition: management implications for aquaculture escapees. Water DOI: 10.3390/w12113186. [Google Scholar]
- Talijančić I., Šegvić-Bubić T., Žužul I., Džoić T., Maršić-Lučić J., Grubišić L. 2019. Interactions between wild gilthead seabream Sparus aurata and tuna farms in the Adriatic Sea: morphological and ecophysiological fish adaptations. Aquac Environ Interact 11: 97–110. [CrossRef] [Google Scholar]
- Talijančić I., Žužul I., Kiridžija V., Šiljić J., Pleadin J., Grubišić L., Šegvić-Bubić T. 2021. Plastic responses of gilthead seabream Sparus aurata to wild and aquaculture pressured environments. Front Mar Sci DOI: 10.3389/fmars.2021.694627 [Google Scholar]
- Toledo-Guedes K., Brito A., Garcia de Leaniz C. 2021. Phenotypic convergence in sea bass (Dicentrarchus labrax) escaping from fish farms: the onset of feralization? Front Mar Sci 8: 674635. [CrossRef] [Google Scholar]
- Van Oosterhout C., Hutchinson W.F., Wilis D.P.M., Shipley P. 2004. MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol 4: 535–538. [CrossRef] [Google Scholar]
- Waples R.S., Hindar K., Karlsson S., Hard J.J. 2016. Evaluating the Ryman-Laikre effect for marine stock enhancement and aquaculture. Curr Zoo. 62: 617–627. [CrossRef] [PubMed] [Google Scholar]
- Yeh F., Yang R., Boyle T. 2002. POPGENE, Version 1.32: A Microsoft Windows-Based Freeware for Population Genetic Analysis. Molecular Biology and Biotechnology Centre, University of Alberta, Edmonton, Canada. http://www.ualberta.ca/~fyeh/info.htm [Google Scholar]
- Yue G. 2014. Recent advances of genome mapping and marker-assisted selection in aquaculture. Fish Fish 15: 376–396. [CrossRef] [Google Scholar]
- Žužul I., Šegvić-Bubić T., Talijančić I., Džoić T., Lepen Pleić I., Beg Paklar G., Ivatek-Šahdan S., Katavić I., Grubišić L. 2019. Spatial connectivity pattern of expanding gilthead seabream populations and its interactions with aquaculture sites: a combined population genetic and physical modelling approach. Sci Rep 9: 1–14. [PubMed] [Google Scholar]
Cite this article as: Žužul I, Grubišić L, Šegvić-Bubić T. 2022. Genetic discrimination of wild versus farmed gilthead sea bream Sparus aurata using microsatellite markers associated with candidate genes. Aquat. Living Resour. 35: 8
All Tables
Information of sampling locations, year and population code, along with the origin and number of individual gilthead seabream that were genetically assayed with 3 microsatellites linked to three candidate genes (cgSST).
Summary statistic for genetic variation of gilthead seabream Sparus aurata in the Adriatic Sea showing the average number of alleles (A), effective number of alleles (Ae), allelic richness (Ar), expected (He) and observed (Ho) heterozygosity, and fixation index (FIS), for 19 neutral SST loci and 3 candidate gene loci (cgSST).
Pairwise FST values based on 3 cgSST loci (below diagonal) and pairwise FST values based on 19 neutral SST loci (above diagonal) among 9 populations of gilthead seabream from the Adriatic Sea, including wild, farmed, farm-associated and wild juvenile populations. Significant FST values are underlined at p < 0.001 (Bonferroni correction). Population codes are explained in Table 1.
All Figures
|
Fig. 1 Sampling locations of wild adult gilthead seabream (WT, Tar Bay; WV, Velebit Channel), wild juveniles (JP, Pantana River; JN, Neretva River), farm-associated adults (AK, Mrdina; AB, Maslinova) and farmed adults (15FC, Budava Bay; 15FI, Mali Ston Bay; 16FF Maslinova Bay) |
| In the text | |
|
Fig. 2 Bayesian STRUCTURE clustering of the gilthead seabream reduced dataset based on (a) 3 cgSST loci and (b) combined 3 cgSST and 19 neutral SST loci, assuming four (K = 4) and five (K = 5) inferred clusters. Vertical coloured lines represent individual admixture coefficients. Black horizontal lines divide individuals from different sampling sites. See Tab. 1 for abbreviated sample names. |
| In the text | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.