| Issue |
Aquat. Living Resour.
Volume 38, 2025
|
|
|---|---|---|
| Article Number | 11 | |
| Number of page(s) | 24 | |
| DOI | https://doi.org/10.1051/alr/2025006 | |
| Published online | 23 June 2025 | |
Review Article
States of development and application of genetic and genomic tools in aquaculture and conservation programs: a guide for strengthening dialogue among practitioners of aquaculture and genetics
1
NOAA Fisheries, Southwest Fisheries Science Center, 8901 La Jolla Shores Drive, La Jolla, California, USA
2
Fisheries and Oceans Canada, Bedford Institute of Oceanography, Dartmouth, Nova Scotia, Canada
3
Ifremer, 29280, Plouzané, France
4
Population Genetics Research Group, Institute of Marine Research, Bergen, Norway
5
Fisheries and Oceans Canada, 200 Kent Street, Ottawa, Ontario, Canada
6
Fisheries and Oceans Canada, Northwest Atlantic Fisheries Centre, St. John's, Newfoundland and Labrador, Canada
7
Aquaculture Solutions, Natural Resources Institute Finland (Luke), Tekniikankatu 1, 33720 Tampere, Finland
8
Natural Resources Institute Finland (Luke), Latokartanonkaari 9, 00790 Helsinki, Finland
* Corresponding author: This email address is being protected from spambots. You need JavaScript enabled to view it.
Received:
24
January
2024
Accepted:
10
April
2025
Abstract
Throughout all stages of fish conservation and aquaculture development, genetic and genomic approaches can be leveraged to enhance understanding of the diversity and complexity of these organisms, including the linkage between phenotype and genotype, and their adaptive and breeding potential. These approaches can inform processes ranging from the initial collection of wild broodstock to the ongoing use of genomic selection on domesticated lines. Due to the diversity in cultured fish species, small and medium enterprises (SMEs) commonly explore new species for culture, or work with species within a narrow regional conservation or commercial focus. These enterprises face obstacles in utilising genetic and genomic approaches due to development and implementation costs, specialised skill set requirements, and infrastructure and labour limitations; yet the benefits often outweigh these challenges. Choosing the best molecular genetic or genomic tools depends on programme goals and species, but small and medium enterprises may miss opportunities to acquire more information through their current approaches, or not realise what may be gained through modest investments in genomic tools. To provide better insight and promote discussion and collaboration between culturists and genomic practitioners, we define and describe five States of development and application of genetic and genomic tools frequently observed in aquaculture and conservation breeding programs. We characterise these tools, their general applications, and how current technologies allow programs to advance to higher States without following a sequential progression, a concept we refer to as “State skipping”. This document outlines the available molecular genetic and genomic tools, but does not cover animal breeding or the science behind it. Similarly, bioeconomic models are not included, although relative economic costs and benefits are highlighted. The technical considerations and limitations of various approaches are reviewed, along with available resources for those seeking further support in exploring genetic and genomic tools in breeding programmes.
Key words: Breeding programs / genetic and genomic approaches / technical considerations / selection / broodstock / small and medium enterprise
Handling Editor: Carlos Saavedra
Catherine M. Purcell and Brendan F. Wringe are considered joint first authors. Equal contributors.
© C.M. Purcell et al., Published by EDP Sciences 2025
 This is an Open Access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This is an Open Access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
1 Introduction
Plant and animal agriculture have been shaped by domestication and directional selection for traits deemed beneficial to humans, dating back at least 13000 years (Diamond, 2002). While many aquaculture species have undergone selection, domestication of aquatic species began much more recently than in terrestrial plants and animals, with the earliest domestication of fish species occurring within the past 2000 years (i.e., the common carp, Cyprinus carpio, in south central Europe by the Romans) to 1000 years (i.e., the goldfish, Carassius auratus, in China; Balon, 2004). Selection methods have evolved from the initial unintentional selection associated with domestication, to phenotypic mass selection, family selection, marker-assisted selection, and now genomic selection (see Boudry et al. (2021) for a review). The adoption in animal and plant breeding of molecular genetic analysis, which we define as the genotyping of a small number of markers to assess variation in individuals or populations, quantify trait heredity, and link markers to specific traits or genes. This contrasts with molecular genomic analysis, which involves genotyping of thousands or more markers to describe genome structure, and explore the interactions of genetic elements and their functions across the genome and their relationship to traits or processes of interest. Genomic approaches have demonstrated objective, quantifiable benefits, both in general (Rexroad et al., 2019) and specifically in aquaculture (Houston et al., 2020). In this paper, we focus on the description of molecular genetic and genomic tools and analyses and their use in providing information on the genetic architecture underlying traits. The adoption of such tools enables an understanding of the composition, diversity, and adaptive and selective potential of a population or strain, as well as a linkage of genetic regions, pathways, and ultimately single genes or variants to traits of interest.
As such, the adoption of genetic and genomic tools is important at all stages of aquaculture development. This can range from descriptions of candidate species and populations, to the initial collection of broodstock, or source material collected annually from the wild, through fully domesticated broodstocks which may be exposed to selective breeding using the most sophisticated genomic selection methods (Fig. 1). Genetic and genomic tools have been successfully used in aquaculture in a number of different settings, including to assist in selection for: disease resistance (Fuji et al., 2007; Moen et al., 2009; Houston et al., 2010; Yáñez et al., 2022; AquaGen's innOva BCWD selected stock (https://aquagen.no)), delayed maturation (Kause et al., 2003; Moghadam et al., 2007), growth rate (Palaiokostas et al., 2013; Tsai et al., 2015; Wang et al., 2017; Garcia et al., 2018; Gutierrez et al., 2018; Yoshida et al., 2018; Yoshida et al., 2019; Joshi et al., 2020; Vu et al., 2021; Wang et al., 2021; Gong et al., 2022; Jerry et al., 2022; Ke et al., 2022; Verbyla et al., 2022), feed conversion efficiency (Besson et al., 2019; Barria et al., 2021; Besson et al., 2022), external appearance (Kause et al., 2004; Colihueque, 2010; Colihueque and Araneda, 2014), product traits, including flesh quality (Quinton et al., 2005; Kause et al., 2011; Nguyen, 2016), and survival (Vehviläinen et al., 2008; Vehviläinen et al., 2010; Vehviläinen et al., 2012).
While initial selection in a species for a few commercially most important traits typically shows large effect (e.g., Neira et al., 2006; Gjedrem et al., 2012), achieving desired results is more difficult for multi-trait selection, and especially when those traits have unfavorable genetic correlations. Moreover, where heritability is on the lower end of the useful heritability spectrum, genomic selection (Meuwissen et al., 2001) can result in greater genetic gain because the accuracy of predicted genetic values increases proportionally higher than for more heritable traits (Rexroad et al., 2019). It is for these traits with low heritability, and also in traits that require lethal measurements from sibs (e.g., disease resistance, carcass traits), where understanding and then applying the tools to target the genetic and genomic architecture of a species or breeding population (i.e., DNA-informed breeding; Peace, 2017) may lead to substantial improvements in the breeding outcome and thus economic benefit (Rexroad et al., 2019).
The genetic and genomic tools developed for commercial aquaculture species can also be used in any breeding effort targeting restocking and conservation (Casey et al., 2016; Waples et al., 2020; Wenne, 2023). For example, in modern conservation programs, genetic and genomic tools are utilised to support the sustainable reproduction of endangered species or populations. In this context, the applications of these tools include defining populations of interest, documenting genetic and genomic characteristics, assisting in the determination and tracking of pedigrees, and monitoring changes in genetic characteristics over generations (e.g., DFO, 2018).
Here, we consider small and medium enterprises (SMEs) to be programs of smaller scale, or independent operations, without pooled investment, coordinated research, or genomic resource development. This scenario is common in species whose suitability for culture is being explored, those who have not been in culture long, species of regional conservation concern, or whose distribution or culture is regionally restricted. It also may apply to more established species in which, despite the existence of genetic and/or genomic tools and information, groups must develop their own resources due to proprietary restrictions, such as when genomic resources are developed by a competitor who may not wish to make the data available. Across all scenarios, the development and application of genomic tools will require financial resources and expert personnel for their development and implementation, as well as infrastructure in which to carry out a breeding programme.
There are many possible obstacles to the adoption of these genetic and genomic approaches, particularly for SMEs with inherently limited infrastructure and research budgets focusing on aquaculture and conservation programmes. These obstacles include the cost of development and implementation of genetic technologies, the availability of specific genetic tools for the species being raised, and the specialised knowledge or expertise required to undertake labouratory, bioinformatics, and analytical work. However, without a shared understanding of the available tools and the overall complexity of their applications, it becomes challenging for SMEs to initiate the necessary internal dialogue with genetic and genomic practitioners to determine their best course of action.
While the financial investment in the approaches described is a significant factor, the potential economic gains should be weighed against the cost of the chosen approach(s) (e.g., Janssen et al., 2018; Fernández Sánchez et al., 2022). Although bioeconomic modeling and economic analysis are crucial, they are not included in this manuscript. Such analyses should be tailored to a specific producer and rely on the most current economic data to be meaningful. Therefore, while costs and benefits may be mentioned, they are presented in general and in relative terms. That said, we will also argue that the use of genetic and genomic tools offers benefits that outweigh the costs (e.g., Abdelrahman et al., 2017; Supple and Shapiro, 2018; Houston et al., 2020; Ghildiyal et al., 2023). In some instances, these tools may even be required by legislation (e.g., https://leap.unep.org/countries/no/national-legislation/regulation-no-1109-quality-parameters-wild-atlantic-salmon-stocks; accessed 26 September 2023; and genetic marking and genetic continent of origin testing required for Atlantic Salmon stocking in U.S. navigable waters and State of Maine waters, D. Bean, NOAA NMFS, pers. comm.).
In light of the considerations above, we explore the application of molecular genetic and genomic tools in both conservation and commercial settings, and introduce a novel five-State system. This system categorises the level of genetic/genomic resources and knowledge that is presently available for candidate aquaculture species, and evaluates their applicability within a breeding program. These States encompass the full spectrum from species for which no genetic data exist and must be developed, to cases where genomic selection is routinely used to select for advantageous traits. Finally, we briefly address the potential use of gene editing. By outlining these States, we aim to facilitate informed discussions between SMEs, aquaculturists, and genetic and genomic practitioners, providing greater context to guide genetic or genomic resource development, as well as the development of breeding objectives.
This review is the product of the International Council for the Exploration of the Seas (ICES; https://www.ices.dk/) Working Group on the Application of Genetics for Fisheries and Aquaculture (WGAGFA), and as such, examples presented tend to focus on species of relevance to ICES member countries. That said, the technologies and techniques discussed are applicable to any species in aquaculture or conservation breeding programs.
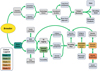 |
Fig. 1 Decision trees that aquaculture program managers must consider to guide development of their operations. These decisions range from choices regarding organism selection, research strategies, and considerations of economic and technological limitations for both commercial and restoration-focused breeding programs. Coloured nodes refer to the States of development and application of genetic and genomic tools, and correspond with the colors and States described in Table 1. |
2 States of genetic method application
Here, we define and describe five States of development and application of genetic and genomic tools for aquaculture and conservation breeding programs. For each State, we characterise the tools used, as well as the types of applications that may be common to that State for smaller-scale commercial aquaculture and conservation-focused programs (summarised in Fig. 2). Where possible, examples are highlighted for both commercial and conservation programs (Tab. 1). The term “State” is purposefully chosen to remove connotations or implications that a program should or must advance from one State to the next; a program may find that the tools and data available at a given State may be adequate for their needs and thus remain at that particular State. In addition, current technologies make it much easier for programs to transition directly to higher States and this is discussed in its own section below.
 |
Fig. 2 The six large panels represent the States of development, the application of genetic and genomic tools, and broad research categories associated with each State. Inset are the relative levels (low, medium, or high − depicted by one, two, or three icons, respectively) of resource investment likely required to address research at each State: sampling or phenotyping labour, breeding program infrastructure (e.g., tanks, labour), genetic or genomic labouratory support (e.g., equipment and specialised labour), and bioinformatic support (e.g., computational and bioinformatics labour). While the requirements for each of these categories of resources may vary widely across different programs at each of the States, the relative levels are intended to depict a minimum level of resource investment that new adopters could expect with research or operations conducted at each of these States. |
States, and state skipping, describing the development and application of genetic and genomic tools in aquaculture and conservation breeding programs, with selected examples under each category as available.
2.1 State 0: No previously developed DNA markers or genomic information
This State refers to species lacking any developed genetic tools or genomic resources (e.g., sequence variants, linkage maps, etc.), either for evaluating wild populations or characterising cultured programs. Programs at this State may collect and rear wild-caught individuals in cages or tanks until they reach market size, or may be in the initial stages of determining whether a species, either for commercial or conservation purposes, is amenable to culture. While many species of interest have now been subjected to study with DNA markers, tripletail (Lobotes surinamensis), serves as an example of a candidate aquaculture species in this State. This fish is of interest for culture in the U.S.A., and wild broodstock have been collected to identify and optimise spawning techniques for the development of commercial culture (Saillant et al., 2021). To date, there are no genetic resources available for tripletail, and no genetic studies have been conducted on this species other than DNA barcoding using a universal mitochondrial marker (i.e. not species-specific) approach (Sirisha et al., 2018). If a culture programme seeks to apply genetic methods for this species, then markers will need to be developed de novo.
For species in State 0, the development of genetic and genomic tools has become considerably easier, owing to advancements in techniques that now make de novo genetic resource development relatively straightforward. This document focuses on examples of these different types of tools and their applications, rather than focusing on the development process itself.
The development of microsatellite markers, and their application to aquaculture was reviewed by Chistiakov et al. (2006); since that time, methods to identify microsatellites and SNPs have advanced considerably. Reduced representation sequencing (e.g., RADseq, and others) has been used to discover SNPs (single nucleotide polymorphisms) (e.g., Bootsma et al., 2020; Chang et al., 2021), microsatellites (e.g., Liu et al., 2021), and haplotypes (e.g., Baetscher et al., 2018; Euclide et al., 2022), across the genome without a reference genome. However, when available, high-quality whole-genome assemblies allow for identification of SNPs and microsatellites using in silico methods (e.g., Marcy-Quay et al., 2023).
When deciding on the type of genetic or genomic marker development to pursue, programmes or species in State 0 should take into account both future objectives and the potential opportunity costs of limiting the number of DNA markers produced, as well as the versatility of the selected molecular tools. Therefore, even for programs that do not anticipate progressing beyond State 2, specifically, individual genetic tagging, we generally recommend developing an annotated reference genome, if feasible. This is due to the advantages it provides including relatively simple development of goal-specific molecular marker panels, as well as serving as the foundation for future efforts such as initiating marker assisted breeding and linking traits and markers to putatively causative genes (e.g., Kess et al., 2019; Kess et al., 2021; Holborn et al., 2022; Lehnert et al., 2023). Leveraging genome information will be discussed in more detail in the section on “State skipping”.
2.2 State 1: Leveraging of population genetic or genomic information
Once a candidate species for aquaculture or conservation has been identified, local regulations generally require permits or licenses for both collection of aquatic organisms from the wild, as well as the aquaculture operations themselves. In many cases, the issuance of collection permits will be contingent upon the applicant demonstrating that the collection will not result in harm to the populations or species. Part of this process may involve the delineation of population structure and determination of effective population sizes (Ne). The delineation of population structure can be accomplished using population genetics or genomics, and this is often the first opportunity to develop and employ genetic or genomic tools and collect genetic information for the species. This information can then be utilised in other States and stages of aquaculture program development.
There are myriad examples of population genetic and genomic studies in aquatic organisms. First based on allozymes, most studies now rely on microsatellite or single nucleotide polymorphisms (SNPs) as markers (see notably Wenne, 2023 for a review about the applications of SNPs in conservation and exploitation of aquatic populations). Whatever the choice of marker, the goal is typically to define population units, determine degree of differentiation between them (e.g., O'Reilly, 2006; Lehnert et al., 2023), and when collection and incorporation into an aquaculture or conservation breeding program is the goal, determine the genetic or genomic diversity within the collection (O'Reilly and Harvie, 2010).
The choice of marker type, the degree of population divergence, and the number of markers used in a study can influence the degree of genetic differentiation that is detectable. While studies historically relied on a handful of markers, advancements in genotyping technologies have made panels of tens to hundreds of microsatellites (e.g., Bradbury et al., 2018) or hundreds to thousands of SNP markers the norm (e.g., Jeffery et al., 2018).
Lehnert et al. (2023) used a weight-of-evidence approach and a combination of genetic and genomic data to identify previously unresolved landscape-scale genetic distinction in Atlantic salmon (Salmo salar). Similarly, using a panel of 101 genome-wide microsatellites, Bradbury et al. (2018) resolved 26 predominantly river-level Atlantic salmon genetic reporting groups across approximately three degrees of latitude in Labrador, Canada, where previous analyses using smaller numbers of SNPs were only able to detect three groups in the same area (Jeffery et al., 2018).
Another example is seen in population genetic studies of lumpfish (Cyclopterus lumpus), a species where both wild-caught translocated and domesticated individuals are used in the aquaculture industry for biological control of sea lice. A study based upon 14 microsatellites indicated that interbreeding between wild and aquaculture escapes would have little impact on the genetic composition of the wild stocks in Norway (Jónsdóttir et al., 2018), while more recent genomic analyses across the Atlantic have revealed fine-scale population structure and temperature adaptation, with the risk of breakdown of local adaptation if introgressed by aquaculture escapees (Jansson et al., 2023; Langille et al., 2023).
Within conservation programs, a common goal is the maintenance of existing genetic characteristics of the species or populations of concern. This generally entails undertaking a population genetic or genomic study to determine metrics such as genetic or allelic diversity, levels of heterozygosity, and effective population size. Theoretical linkages can then be made to predict the risk to the species or population of genetic impact from inbreeding and drift over time, among other concerns (e.g., O'Reilly and Harvie, 2010). In conservation scenarios where focal population(s) has(have) been extirpated and reintroduction is desired, it is generally recommended to identify and use source populations that are genetically − and presumably adaptively − similar. These can be identified by screening nearby existing populations and, if they exist, comparing them to focal population samples (e.g., Anderson et al., 2014). An associated approach, genetic rescue, may be used for populations where reductions in genetic diversity are reducing the fitness and resilience of the population. In this scenario, individuals are intentionally translocated into the imperiled population to restore genetic variation and adaptive potential (Kovach et al., 2022). While difficult to predict, the outcome of genetic rescue may rely in part on the effective population sizes of the recipient and donor populations and their genetic divergence, metrics which can be measured using population genetics (Wells et al., 2019).
2.3 State 2: Individual genetic tagging
One of the most immediately informative genetic approaches to utilise for a breeding program is individual genetic tagging. Individual genetic tagging refers to the use of DNA markers to unambiguously identify an individual, link an individual to genetic information pertaining to family relationships (e.g., parentage and pedigree analyses) (Vandeputte and Haffray, 2014; Liu et al., 2016; Holman et al., 2017; Weng et al., 2021), and/or to trace the individual back to their breeding program, or origin (e.g., farmed versus wild, and/or location) (Norris et al., 2000; Chistiakov et al., 2006). Within selective breeding programs, this can allow (previously collected) phenotypic data to be linked to an individual, or to their relatives, through parental assignment and pedigree reconstruction. This can enable higher resolution tracking of phenotypic heritability and inheritance through breeding lines, as well as management of ancestry and inbreeding within conservation programs (Holborn et al., 2025). Detection of linkages between DNA markers and genomic regions is discussed in more detail in States 3, 4 and 5 below.
Both parental assignment and pedigree reconstruction can be used to manage the level of inbreeding and genetic diversity in both commercial and conservation breeding programs across generations (Meuwissen and Sonesson, 2004; Vandeputte and Haffray, 2014; Wellmann et al., 2014; Gebregiwergis et al., 2020; Gautason et al., 2022). Given the importance of genetic diversity in maintaining the adaptive capacity of populations to respond to novel challenges in the environment (Tringali and Bert, 1998), in providing the variation upon which long-term selection goals in breeding programs rely on (D'Ambrosio et al., 2019), and in avoiding detrimental impacts due to inbreeding associated with a loss of genetic diversity (Kincaid, 1976), mitigating the loss of genetic diversity in breeding programs is a high priority.
Where pedigree data do not exist, genetic kinship data available across generations can be used for parental assignment (Lacy, 2012) and pedigree reconstruction (Mendes et al., 2022). Parental assignment or pedigree data can be helpful in exploring spawning dynamics and assessing individual spawning success for naturally spawning species in culture settings (Herlin et al., 2007; Herlin et al., 2008; Horreo et al., 2008). These analyses may also assist in tracing physical traits (due to genetic or non-genetic factors) back to individual broodstock. This type of application can be immensely helpful to rapidly inform breeding designs and culturing approaches.
In the California yellowtail (Seriola dorsalis), for example, parentage analyses using a small number of microsatellites (developed for other Seriola species) revealed that only one, or occasionally two, female broodfish contributed to individual spawning events and dominated spawning seasons regardless of female abundance in the tank (Schmidt et al., 2021). This discovery informed experimental approaches which used smaller breeding groups spread among an increased number of smaller spawning tanks. Parental assignment was also used to identify females contributing low-quality eggs, which informed decisions to bring in additional broodstock.
In the White Abalone (Haliotis sorenseni) Recovery Program (a collabourative effort with the University of California, Davis, NOAA Fisheries Southwest Fisheries Science Center, and other partners), parentage and pedigree analyses, using microsatellite markers, informed crosses of gametes for the captive breeding program to maximise genetic diversity in this endangered species. As demonstrated above, determination of broodstock breeding dynamics and quantification of the extent of unequal reproductive success can help guide targeted mitigations that include modifications to spawning groups, alterations of breeding tank designs, replenishment or rotation of broodstock individuals or populations, and help determine whether other approaches such as artificial fertilization or cryopreservation are needed.
Supplementation or conservation program effectiveness may also be evaluated using genetic marking and pedigree reconstruction (Araki et al., 2007; Horn et al., 2023). In order for a conservation program to be successful, the individuals removed from the wild population to be bred in captivity must produce at least as many adult offspring as they would have had they been left to reproduce in the wild. Whether the program meets this objective can be evaluated by genotyping the parents in captivity and subsequently sampling and genotyping the next generation of adults and conducting genetic parentage assignments. This methodology may also reveal if the parents in captivity contribute to the next generation to such an extent that it ultimately reduces the effective population size and thus the genetic variation in wild populations (Ryman and Laikre, 1991).
For supplementation programs, providing harvest opportunities may also be among the objectives. Such is the case for Canada's West Coast Salmonid Enhancement Program, which includes among its goals both rebuilding vulnerable populations and providing harvest opportunities. The Salmonid Enhancement Program uses parentage-based tagging to track fish captured in the fishery back to their hatchery (and thus population) of origin. While technically not a breeding goal, genetic tagging can also be used for product traceability in production systems, and traceability to the producer or cage facility is a legal requirement for aquaculture in some jurisdictions (Håstein et al., 2001; Espiñeira and Vieites, 2016; Holman et al., 2017).
From a conservation perspective, the goals of captive breeding are avoiding loss of genetic variability and inbreeding, and avoiding genetic drift and accidental selection, while other program-specific targets such as retention of rare alleles or maintenance of a target effective population size may also exist (Rollinson et al., 2014; Waters et al., 2015; Attard et al., 2016; Marshall et al., 2022). Additionally, it is important to monitor both the genetic character of reintroduced populations, and the effect of those reintroduced individuals on the genetic character of the wild remnants of the species/population (Russello and Amato, 2004; Christie et al., 2012; Christie et al., 2013; Waters et al., 2015; Waters et al., 2016; Waters et al., 2018; Auld et al., 2021; Hagen et al., 2021; Marshall et al., 2022), particularly to avoid the Ryman–Laikre effect (Ryman and Laikre, 1991), where hatchery fish increase the number of spawning individuals while simultaneously decreasing the effective population size (Morvezen et al., 2016).
As an example, genetic approaches are used to determine the origin of Norwegian Atlantic salmon. Due to declines in natural populations, Atlantic salmon is being stocked for conservation purposes in multiple areas of their natural range. Genetic screening of wild-caught broodstock can be an effective tool for improving the accuracy of such programs. From a 7000 SNP array, a set of 59 SNPs have been identified with the purpose of separating Norwegian Atlantic salmon individuals of farmed vs. wild origin (Karlsson et al., 2011; Karlsson et al., 2014). Introgression of farmed salmon has been documented throughout Norway (Karlsson et al., 2016), leading not only to genetic but also phenotypic and phenological alterations in introgressed individuals (Bolstad et al., 2017; Bolstad et al., 2021; Besnier et al., 2022). It has also been documented that farmed ancestry has been unintentionally selected for in the supplementary stocking of Atlantic salmon (Hagen et al., 2019). As a result, genetic screening for introgressed individuals in broodstocks used for conservation breeding is now a routine practice in Norway.
As demonstrated in the examples above, it is still possible to use genetic tools such as microsatellites or sets of SNP markers for the applications described under this State. However, some breeding goals may benefit from the adoption of genomic technologies due to the higher potential for precision they have for estimating population parameters (Supple and Shapiro, 2018). In this State, the analytical and bioinformatic support required for these applications is not extensive, but will depend on the scale of the undertaking and the types of analyses required for the desired project.
2.4 State 3: Linkages between phenotypes and genotypes
In State 3, genotyping is used to establish associations between DNA markers and phenotypes, and these linkages are used to inform the breeding process or other aspects of the program. Implementation or description of breeding programs is beyond the scope of this paper. The focus in this State is on targeting simple traits with Mendelian inheritance or phenotypes associated with genes — and their associated DNA markers — of major effect, whereas polygenic associations and genomic imputation are discussed in State 4.
Approaches to developing linkages to phenotypes include Quantitative Trait Loci (QTL) analysis and Genome-Wide Association Studies (GWAS). QTL analysis identifies genetic DNA markers associated with the traits, primarily within pedigree studies. On the other hand, GWAS, a more recent evolution of QTL analysis, identifies QTLs using population-scale sequencing and maps phenotypes to genotypes. While linkage maps may be produced through various forms of reduced representation sequencing to obtain markers across the genome, a reference genome may also be used for this purpose. Developments of linkage associations at this level allows programs to use marker assisted selection (MAS) to identify and screen individuals at target loci associated with traits of interest. Marker assisted selection and/or application of genetic screening for these traits offers a rapid approach to make improvements in the breeding program or inform breeding decisions (Meuwissen et al., 2001).
Widespread examples exist of the use of genomic information described in State 3 in culture or study of aquatic organisms, but frequent applications include identification of genetic sex (Einfeldt et al., 2021; Holborn et al., 2022), improvements in disease resistance (Holborn et al., 2019; Holborn et al., 2020), and determining the degree of introgression by aquaculture escapees in fish destined for breeding programs (Liu et al., 2017; Bradbury et al., 2022). For example, the reference genome developed for California yellowtail (Seriola dorsalis) was used in conjunction with a GWAS on resequenced (i.e. low-coverage whole-genome sequencing) sexed individuals to identify a genomic region associated with sex (Purcell et al., 2018). This approach identified both a genomic region significantly associated with sex, but also an insertion/deletion within that region. PCR primers spanning this region were used to amplify and visualise fragments on an agarose gel, enabling sex identification in brood and offspring fish based on the banding pattern (e.g. presence or absence, with positive control). This method has become a valuable tool for a species lacking external sexual dimorphism (Purcell et al., 2018).
At the NOAA Fisheries, Southwest Fisheries Science Center, in collabouration with Iowa State University, a similar approach is currently underway for the endangered white abalone (Haliotis sorenseni) to enhance the breeding programme. A recently assembled chromosomal-scale reference genome for this species is being used to inform a GWAS using genotyping-by-sequencing (GBS) data from sexed white abalone specimens. One highly significant genomic region associated with phenotypic sex has been identified, and genetic variants are currently being assessed and screened (as of manuscript preparation) to determine if a sex-specific marker may be developed. The development of a sex-specific marker for this species would be tremendously important to the breeding programme, as it would allow for the identification of potential broodstock without the need for extensive handling of wild abalone to determine sex. A simple swab sample followed by genotyping would be an effective method for these animals with relatively limited dispersal. Additionally, this marker could support outplanting efforts by ensuring mixed-sex groups of juvenile white abalone or facilitating the matching of known-sex individuals with wild populations in the region.
In Atlantic salmon, outbreaks of infectious pancreatic necrosis virus (IPNV) were a major concern to the aquaculture industry. Research groups identified a single major QTL that explained 80 to 100% of genetic variation in resistance to IPNV. Marker assisted selection, utilising a SNP-based genetic screening for the favorable, resistant, and dominant allele, was rapidly adopted by salmon breeding programs to help prevent further outbreaks of this virus (Houston et al., 2012).
Similarly, disease outbreak of Ostreid herpesvirus 1 (OsHV-1) has greatly affected aquaculture of Pacific oysters (Crassostrea gigas). A QTL on chromosome 8 of the Pacific oyster genome was determined to be associated with improved survival (13% phenotypic variance) to mortality events in Tomales Bay, California, USA, where OsHV-1 is endemic. Marker-assisted selection for this QTL resulted in 47% higher survival in breeding values for families undergoing MAS than based solely on pedigree selection (Divilov et al., 2023).
Developing and applying genotype-phenotype linkages may also be important for restoration-focused culture. A good example is the European flat oyster, Ostrea edulis, which is currently a species of limited interest in commercial aquaculture, particularly when compared with the Pacific oyster. However, there is an increasing interest in oyster bed restoration, particularly in northern Europe where populations have been severely depleted, or become extinct due to overfishing, parasites and habitat degradation (see https://noraeurope.eu/). Genomic tools and knowledge have been developed for European flat oyster, and these currently include several low- to medium-density SNP panels (Lapègue et al., 2014; Gutierrez et al., 2017), RAD-Seq data, three independently developed chromosome-level assemblies, and low-coverage whole-genome sequencing (Bean et al., 2022; Boutet et al., 2022; Gundappa et al., 2022; Li et al., 2023). While most studies have been dedicated to population genomics aimed at studying population structure and identifying signatures of local adaptation (e.g., Vera et al., 2019; Lapègue et al., 2023), GWAS has also been applied in hatchery-produced offspring to target traits of interest for aquaculture (Peñaloza et al., 2022). In particular, molecular breeding to improve resistance against bonamiosis (an infection caused by a protozoan parasite that impacts wild and cultured populations) is a major objective in this species (Pouvreau et al., 2023). However, the development of selective breeding programs remains limited for O. edulis, notably due to the small-scale nature of the aquaculture industry, and to the constraints related to seed supply for restoration projects (Colsoul et al., 2021; Zu Ermgassen et al., 2023).
Linking single traits to DNA markers is also of consideration for conservation. Determination of what constitutes a ‘species’ is foundational to conservation as well as endangered species legislation, especially given that most programs and legislation consider subunits below the level accepted as biological species. For instance, in determining designatable units, the Committee on the Status of Endangered Wildlife in Canada assesses the proposed group's discreteness and evolutionary significance (Lehnert et al., 2023; Lehnert et al., 2023). This is similar to the U.S. evolutionarily significant unit (ESU) being based on ‘reproductive isolation’ and ‘evolutionary legacy’ (Waples et al., 2022). These determinations are generally accomplished through investigation of population structure using panels of typically neutral markers (e.g., Lehnert et al., 2023). However, adaptive diversity at single markers exists within identified conservation units, and the appropriateness of considering designations based on variants at these markers has been debated (Waples et al., 2022).
Within salmonids, for example, variants in single genes have been detected that have a major influence on important life history traits. In Atlantic salmon, variants of the genes six6 and vgll3 show pronounced effects on age at maturity in some genetic and habitat backgrounds (Barson et al., 2015; Sinclair-Waters et al., 2020; Kess et al., 2024). In Pacific salmon, six6 has also been associated with age at maturity in Steelhead (Oncorhynchus mykiss) and Sockeye salmon (O. nerka), but no association was found in Chinook (O. tshawytscha) or Coho salmon (O. kisutch) (Waters et al., 2021). Of particular note for conservation programs are the links that have been detected between an approximately 200 Kb region of the Steelhead and Chinook salmon genomes between the genes GREB1L and ROCK1 (Hess et al., 2016; Narum et al., 2018). Variants at this region are associated with the propensity to exhibit ‘early’ or ‘late’ migration, where the migration type enables access to spawning habitats that are inaccessible under different flow/temperature conditions (e.g., sandbars can block river mouths during low flows). Despite being loci of large effect, the presence of both variants in a number of populations suggests that this diversity is being preserved through ongoing selection. The importance of selection in maintaining diversity is also demonstrated by changes in allele frequencies following interbreeding when habitat that was previously inaccessible to one or the other type is created through anthropogenic alteration. Waples et al. (2022) discuss loci of large effect in Pacific salmon in relation to conservation and the definition of conservation units.
The above section provides a broad spectrum of scenarios for which linkages between DNA markers and traits have been developed and utilised in both commercial and conservation focused programs. In this State, while it may be possible in some cases to utilise existing DNA markers for many species, applications in this State may require additional steps such as developing a reference genome and using higher-resolution genotyping or sequencing for GWAS studies. A greater degree of analytical, computational and bioinformatic support will be required to accomplish these goals, but requirements for ongoing support will be largely determined by the method used to link the marker and trait.
In conclusion, it is important to note that the overwhelming majority of markers available in marker panels are not causal. Thus, while powerful, all approaches described in Stage 3 are dependent on the strength of the linkage between markers and phenotype, and the persistence of variation at markers linked to the phenotype. For linked, non-causal markers, a recombination-driven breakdown of linkage disequilibrium over generations (that is, a trend towards an independent segregation of genetic marker and causal variant), or, conversely, the loss of heterozygosity due to marker-driven selection, will make specific linked markers lose their information content, creating the need to regularly identify high-information-content markers. The level of polygenicity of the phenotype or phenotypes will influence the amount of variance any marker will be able to explain, with smaller proportions of variance attributable to any specific marker with greater polygenicity of a trait. These caveats are relevant to any genetic program that is, or has been, at a stage corresponding to Stage 3. However, this challenge is being overcome through statistical approaches that model combined sub-significant contributions of alleles to phenotypes, and ongoing reductions in sequencing cost and increases in data informativeness that have allowed the broader genotyping across large, phenotypically diverse sample sets (i.e., State 4).
2.5 State 4: Genomic selection and/or genomic imputation and prediction using family- or pedigree-based selection
In State 4, genomic selection and/or genomic imputation and prediction are applied using population- or pedigree-based selection methods. In this State, many species will have been bred in captivity and subjected to selection for multiple generations. However, genomic prediction methods are now being increasingly applied to wild populations of conservation interest as well. The common theme unifying these approaches is the leveraging of existing genomic information, typically including an annotated reference genome, to predict unobserved data. This can include traits of interest, signatures of environmental association, or the allelic state at a specific locus.
Having a high-quality genome for a species of interest also creates opportunities to generate genome-level data for individuals at considerably reduced costs through imputation or low-coverage whole genome sequencing (Lou et al., 2021). When high-density marker maps are available for the parents of the samples under investigation, it becomes possible to sequence fewer loci and then impute to the whole genome (Kijas et al., 2017). Imputation methods use missing genotype information at the pedigree or population level to assign unobserved or low-confidence genotypes, thereby reducing the amount of missing data (Marchini and Howie, 2010).
Alternatively, genotype likelihood approaches use the combined information across sequenced individuals to infer population genetic parameters (Kim et al., 2011). These methods have been generally found to outperform reduced-representation and SNP array-based approaches in terms of informativeness for detected trait-associated loci (Homburger et al., 2019; Jorsboe and Albrechtsen, 2022), and in accurately inferring population divergence (Szarmach et al., 2021). In recent years, whole-genome sequencing approaches that use low-coverage data, along with imputation or error correction approaches, have been increasingly applied in both aquaculture (Tsai et al., 2017; Gundappa et al., 2023; Kriaridou et al., 2023) and wild population studies (Kess et al., 2024), when a reference genome is available to increase the number of informative markers for genetic analyses. Imputation-based whole genome sequencing (WGS) approaches have been applied to Atlantic salmon in both aquaculture and wild population settings for marker discovery and GWAS (Kess et al., 2024). Similarly, low-coverage WGS and genotype likelihood methods have been applied to various wild species of management interest, including Atlantic silversides (Menidia menidia) (Therkildsen et al., 2019), European eel (Anguilla anguilla) (Enbody et al., 2021), northern sand lance (Ammodytes dubius) (Jones et al., 2023), and cunner (Tautogolabrus adspersus) (Nugent et al., 2023), to uncover population differentiation relevant to species management. These methods represent the forefront of WGS approaches, optimising the trade-off between marker and individual sample number, while providing robust estimates of genomic diversity, population structure, and trait association. However, reference population divergence, as well as local population structure and linkage, can bias imputation accuracy (Lou et al., 2021). In cases of low divergence and linkage, or high divergence from a reference population, genotype likelihood approaches may serve as a validation step (e.g., Kess et al., 2024).
Genomic predictions are increasingly being used in both wild and aquaculture studies, to infer unobserved phenotypes from observed genomic data. With advances in high-throughput sequencing (HTS), genomic selection (Meuwissen et al., 2001) has emerged as a powerful tool in aquaculture to enhance selective breeding programs and improve trait outcomes. Genomic selection relies on the analysis of a large number of markers distributed genome-wide that (ideally) captures all relevant genetic variance, in order to estimate genetic potential or the genomic estimated breeding value (GEBV), of individuals for various traits, including traits with complex genetic architectures. These methods have been used for prediction and trait improvement across numerous aquaculture species (reviewed in Song and Hu, 2022). The automation of this process is also underway, with the development of reference genotype panels for many aquaculture species, as well as web-based infrastructure to support these analyses (Zeng et al., 2022).
Machine learning approaches are now also being applied to better account for the non-linearity of interactions among trait-associated loci (Brieuc et al., 2018), and have shown improvements over traditional additive genetic models under certain genetic architectures (Azodi et al., 2019; Abdollahi-Arpanahi et al., 2020). Although not universal solutions, they have demonstrated success, and therefore, careful consideration is warranted when applying these approaches.
In wild populations, genomic vulnerability approaches provide a methodologically similar approach to using genomic information for predicting phenotypes (Bay et al., 2018; Capblancq et al., 2020), but instead phenotypic information is inferred indirectly via environmental or climatic data, which are assumed to reflect local adaptation (Hoban et al., 2016). Polygenic score approaches have been applied in this context to predict individual environmental associations in species such as the eastern oyster (C. virginica) (Bernatchez et al., 2019) and sea cucumber (Parastichopus californicus) (Xuereb et al., 2018). Additionally, genomic vulnerability estimations of future climate impacts have been conducted in Arctic charr (Salvelinus alpinus) (Layton et al., 2021), marine invertebrates (cape urchin (Parechinus angulosus), common shore crab (Cyclograpsus punctatus), and granular limpet (Scutellastra granularis) (Nielsen et al., 2021)), an additional seaweed (Phyllospora comosa) (Wood et al., 2021) and eelgrass (Zostera marina) (Jeffery et al., 2024). Likewise, all-cause decline rate risk has been estimated at the river level in Atlantic salmon (Lehnert et al., 2019), indicating that genomic prediction methods may offer utility for conservation planning and prioritisation.
2.6 State 5: Gene editing
Gene editing and genetic engineering are rapidly evolving technologies, which warrant consideration as they may see regular application in the future. While the advanced technologies used in genome editing may seem less accessible for SMEs, there could be instances where the benefits of gene editing or genetic engineering outweigh the associated costs or complexities. However, at least one aquaculture genetic/genomic service provider has already advertised gene editing as a service (e.g., https://aquatechcenter.com/services/genome-editing/), which could help make these technologies more accessible to smaller-scale SMEs.
One area of particular interest for smaller SMEs may include gene editing approaches aimed at developing sterile offspring, which could be used in conjunction with other selective breeding goals. This could be especially beneficial in regions where obtaining permits for the use of domesticated lines in ocean environments could be challenging, particularly without additional measures to minimise potential impacts on wild conspecifics in the event of escapes (Wong and Zohar, 2015; Güralp et al., 2020; Kleppe et al., 2022).
However, the acceptance of gene editing or genetic engineering approaches by the public, as well as the legislation governing their use in commercial, and especially conservation programs, varies considerably across the world (e.g., Qin and Brown, 2006; Dietrich et al., 2022). Therefore, it is essential to investigate local legislation and public acceptance before considering their use in a program.
There are multiple methods for performing gene editing, with Zinc fingers (ZFN) (Miller et al., 1985; Klug, 2010; Urnov et al., 2010) and TALENs (transcription activator-like effector nucleases) (Joung and Sander, 2013) being the earliest technologies developed. Today these techniques are considered to be both time consuming and expensive. The CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats)/Cas technology, first presented in 2012, is a simpler, cost-effective and efficient method for targeted gene editing, and can be used in all organisms and cells (Doudna and Charpentier, 2014). Compared to ZFN and TALENs, the CRISPR/Cas technology was identified early as a promising tool (Gaj et al., 2013). However, all three techniques are based upon the same principle: a protein that cuts DNA in a targeted place.
Compared to traditional genetic engineering methods, where whole genes (often from other organisms) are randomly inserted into the genetic material of the target organism, gene editing enables precise modification of genes already present in the organism, to inactivate (knockout) genes or genetic sequences, or to insert (knock-in) genetic material at specific locations of the genome. Thus, a prerequisite for editing genes with CRISPR/Cas is knowing the sequence of the specific piece of DNA to be edited, as well as the complete genome sequence in order to reduce the potential for off-target editing and effects. In this way, knock-in gene editing can also be used to transfer genes from other organisms to specific sections of the genome, so-called transgene knock-ins. Additionally, CRISPR technology has the potential to switch off genes without altering the genetic code, enabling epigenomic alteration (Nuñez et al., 2021), although the full potential for CRISPR-based epigenetic editing is still unknown.
As a potential solution for hindering gene flow from aquaculture escapees to wild conspecifics, the CRISPR/Cas can be used to render aquaculture fish sterile. This can be accomplished by inhibiting the function of proteins that are important for germ cell development and/or survival (Wong and Zohar, 2015), which has been demonstrated in Atlantic salmon (Wargelius et al., 2016; Güralp et al., 2020; Kleppe et al., 2022). There are also several examples where CRISPR/Cas-induced knockout of genes have been shown to improve commercially important traits, such as growth, pigmentation, disease resistance and production of omega-3 fatty acids (see Roy et al., 2022). Recently, tiger puffer (Takifugu rubripe) and red sea bream (Pagrus major) (Kishimoto et al., 2018), which were edited with CRISPR/Cas for enhanced growth, were approved for the Japanese market.
The discovery of the CRISPR/Cas system has also begun to supplant the use of random transgenesis techniques (a subset of genetic engineering). There is a relatively long history of developing and producing transgenic fish in aquaculture through various random transgenesis methods. The first transgenic aquaculture fish were produced over 35 years ago (Devlin et al., 2006), and since then, over 35 species have undergone transgenesis (Devlin et al., 2015), with the AquaAdvantage Atlantic salmon being one of the most well-known examples.
Through traditional gene modification methods transgenes have been introduced into aquaculture species to express desired traits, such as improved disease resistance, altered metabolism, and increased growth (Devlin et al., 2006). CRISPR-edited transgenes have also been produced to enhance disease resistance (Simora et al., 2020), and current efforts are directed towards identifying genes or causative mutations in Pacific salmon related to sea lice resistance (Robinson et al., 2023). If such genes are identified, they could potentially be transferred to Atlantic salmon, a species highly susceptible to sea lice, using the CRISPR/Cas technology.
The transition from using random transgenesis techniques to achieving transgenesis through CRISPR/Cas is also evident in the ornamental fish trade and supporting commercial culture systems. The incorporation of transgenes that express different fluorescent colour proteins, producing novel colour phenotypes, has been conducted through random transgenesis techniques for several species including the Black tetra (Gymnocorymbus ternetzi) and Zebrafish (Danio rerio) (DFO, 2018a; 2019, 2020a, b) (DFO, 2018; DFO, 2019; DFO, 2020; DFO, 2020).
More recently, CRISPR/Cas has been used to introduce the same genes and achieve similar colour phenotypes in Siamese fighting fish (Betta splendens) (DFO, 2021) and a species of Barb (Puntigrus tetrazona) (Dietrich et al., 2022). The single gene to single trait nature of these modifications make the development of these types of ornamental fish relatively straightforward. As these technologies advance, the development of fish that incorporate more sites of modification, and thus greater complexity in trait modification, is likely.
Although few commercial CRISPR-edited species are available for human consumption at present (e.g., Kishimoto et al., 2018), this number may increase because regulations regarding their use are being evaluated in multiple geographic regions. In parallel, more commercial entities are increasing their research efforts related to this powerful technique.
The commercial applications of gene editing are clearer than its use in conservation programs. Moreover, implications to legal conservation status notwithstanding, public and legislative acceptance is more likely for gene editing in commercial settings than for conservation purposes, particularly when the program will result in the release of gene-edited individuals into the wild.
That said, the use of gene editing in conservation programs has been proposed, though primarily in theory. For instance, a scenario could be envisioned where changes to a single region of the genome results in increased immunity to a disease agent that is endangering a species. Alternatively, gene editing could be used in species where genetic rescue is needed, allowing for the identification and targeting of lethal recessive alleles in a genome where genetic diversity has largely been lost and genetic drift has resulted in detrimental effects on the remaining individuals (Segelbacher et al., 2022).
Phelps et al. (2020), outline additional potential applications of gene editing in conservation, ranging from simpler applications (e.g., inducing neutral changes to a single base pair for marking) to significantly more complex and controversial (e.g., utilising synthetic biology to recreate genomes and species that have been lost through extinction).
2.7 State Skipping
In many ways the States presented here are a product of the history of the development of genetic and, subsequently, genomic tools in non-model species. While it is understood that aquaculture has been practiced for thousands of years, the broad adoption of selective breeding and expansion of aquaculture programs began during the latter half of the 20th century. This time period also coincided with the early development and application of genetic tools in non-model species.
A prime example is Atlantic salmon; collections of wild salmon in the 1970s were used to form large-scale family-based breeding programs in Norway (State 0; Gjedrem, 1985). Around this time, descriptions of genetic variation and population genetic studies began to emerge, including hatchery-reared populations (State 1; e.g., McKenzie and Paim, 1969; Moller, 1970; Payne, 1974; Davidson et al., 1989). This was followed by the development of DNA markers for individual and population identification (State 2; e.g., Davidson et al., 1989; Taggart and Ferguson, 1990), the linkage of traits of interest to gene regions (State 3; e.g., Reid et al., 2005), and more recently, the use of genome-scale data in breeding programs (State 4; e.g., D'Agaro et al., 2021) and the development of transgenic lines and gene-edited variants (State 5; e.g., Wargelius et al., 2016; Güralp et al., 2020; Kleppe et al., 2022). However, this transition through stages has been more a consequence of scientific advancements and technology than a deliberate process.
Due to the relative ease with which a de novo, chromosome-level, fully annotated genome can now be produced − from which most commonly used genetic and genomic tools can be developed − new programmes can now rapidly begin at any desired State, effectively skipping those States below the chosen State. State skipping, as we have termed this, has advantages in terms of cost (developing only tools needed), efficiency (tools optimally designed to the problem at hand), and time (application of tools nearly immediately; optimised tools enable faster outcomes). As such, we would recommend that new programs seek to develop genomic resources of broad utility, such as whole-genome assemblies, that will allow for the development of tailored tools. At the same time, developing program goals (e.g., testing for the maintenance of genetic diversity in a conservation program, undertaking genomic selection in an aquaculture strain, etc.) will facilitate conversations with experts to help determine the best set of tools to develop given the goal.
Put another way, the advances in genotyping technology and the reduction in genotyping per-base sequencing costs mean that a candidate species can be moved from State 0 to practically any State of a researcher or SME's choosing with relative ease. For example, cunner is a temperate reef fish native to the northwest Atlantic, that is currently being considered as a candidate species to be used as cleaner fish in Atlantic salmon aquaculture in Atlantic Canada, and for which, until recently, few genetic tools existed (Costa et al., 2016; Monk et al., 2016; Chen, 2020).
Nugent et al. (2023), describe the production and annotation of a chromosome-level genome assembly for this species, as well as the characterisation of genetic population structure in Atlantic Canada. This genome will allow the development of many diverse genetic and genomic tools (e.g., microsatellites, SNPs, copy number variants, etc.). Because the genome is annotated, functional relationships may be determined between traits of interest and DNA markers. Annotated reference genomes can improve the quality of genomic tools by allowing users to avoid repetitive genomic elements and evenly distribute markers across the genome. Moreover, an annotated reference genome allows for the transfer of data and information between projects or groups by referencing standardised genomic coordinates and names for genetic loci.
Finally, comparisons can be made between annotated reference genomes of different species to identify loci or ecologically relevant genes that have been identified as being important in a given species, habitat, or context. An excellent example of the use of a de novo generated annotated genome alongside data from another project is described in Gao et al. (2023). These researchers outline the development of a 50K SNP array designed for North American Atlantic salmon based on SNPs identified in the North American salmon genome they developed, with the European salmon genome's annotation used to approximate the locations of genic regions.
A schematic example of how an annotated genome can be used to develop and apply genomic tools in a breeding program is shown in Figure 3. In this figure the genome is used to enable: marker discovery and selection based on population genotypes and putatively causative genes (blue arrows); changes in population phenotype and genotype as the result of genetically informed selection (orange arrows); the identification of putatively causative genes (garnet arrows); and changes in genotype and phenotype as a result of gene editing (fuchsia arrows).
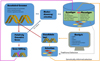 |
Fig. 3 Schematic illustration of the utility of an annotated, whole genome in aquaculture or conservation breeding programs. Solid arrows represent directionality of information or activity with colours denoting common pathways. The grey dashed line, representing traditional selection, is included to reinforce that it operates primarily based on phenotypes and that it can occur while genomic tools are being developed. |
3 Considerations and constraints of genetic and genomic methods
The widespread adoption of genetic methods is constrained by economic and practical considerations. The economic considerations are centered on the overall costs associated with the use of these approaches, and whether the expected outcome or improvement will result in a net economic gain or reaching a conservation goal. These types of considerations and analyses should be specific to the producer and rely on the most current economic data to be meaningful. The practical considerations range from what State the species of interest currently is in, to availability of all ancillary genomic (such as bioinformatic and analysis pipelines, data storage and management), phenotyping (e.g., measurement equipment, personnel, etc.) and facility (e.g., experimental tanks or cages, separate production and selected lines, etc.) resources required to actually translate raw genotype and phenotypic data into actionable breeding or conservation information.
Expense considerations will need to take into account the total cost of genetic/genomic approaches, including both genotypic and phenotypic data collection, data analysis, and data storage, on top of the actual cost of genotyping, animal rearing and husbandry. Part of this consideration should be the development of a breeding objective which should note the traits that influence income, expenses, or both; where the goal is to improve multiple traits, traits should be prioritised based on economic importance (Kankainen et al., 2016; Lhorente et al., 2019). It is worth noting that any cost–benefit analysis regarding the adoption of genetic and/or genomic tools will depend on the species and the technological stage at which the work is conducted. For instance, moving from a high-throughput sequencing approach to an off-the-shelf genotyping solution, or shifting from a dense genome-wide marker set to a low-density marker set with genome-wide imputation, could substantially decrease costs and thus substantially change medium- and long-term cost-benefit projections.
Technical constraints, especially regarding data and sample collection and processing, may hamper the ability of some programs to State Skip. Depending on the specific goals being pursued, different types, and different quantities of data will need to be collected and handled, starting from tissue sample collection for genotyping, up to full phenotypic and genotypic characterisation of hundreds or thousands of samples, potentially over multiple generations. For applications in State 1 and 2, the infrastructure requirements are generally lower as the focus is on sampling wild individuals or broodstock and offspring. Tissue sampling tools and preservation methods are typically inexpensive; methods are available to preserve tissue samples for DNA extraction at room temperature, although in some cases, DNA storage may require freezing, including flash-freezing (e.g., liquid nitrogen). The best approach will depend on the species and on the genetic application. In most instances, staff can be easily trained to collect tissue samples for DNA extraction and processing. However, careful record keeping is essential to match genetic samples back to individual organisms (e.g., utilising physical tags) or families, and phenotype and genotype data.
Beginning in State 3, and more so in State 4, a combination of trained staff and infrastructure investment in phenomics technologies, which allow for the collection of large amounts of phenotypic data, will be necessary to accurately phenotype the animals, depending on the number and complexity of phenotypes. The importance of precise and standardised phenotyping approaches for any State targeting traits cannot be overstated. To a large extent, the success in associating traits to genotypes will depend on the ability to reliably collect accurate phenotypic information. In State 3, phenotypes are often easier to collect (e.g., sex based on gonads, fish length, presence of deformities), but become more complex in State 4 (e.g., fillet yield, color, lipid content, feed conversion rates, disease/parasite resistance), and require not only more highly trained staff, but may also require specialised equipment and infrastructure for measuring some traits. As mentioned above, applications in State 3 and State 4 also require a large number of individuals to be phenotyped and genotyped, and as trait complexity or the number of targeted traits increases, a greater number of samples are needed for analyses. The time, labour, and (if needed) equipment necessary to phenotype large numbers of animals (and repeatedly phenotype in successive generations − as is the case in State 4) should be considered. Often in State 3 and State 4, family and/or selected lines also need to be maintained, which increases the infrastructure required to maintain these populations within the aquaculture program. State 5 has similar infrastructure requirements as State 4; however, the intensiveness of phenotyping will depend on the specific target(s) of gene editing. In addition, State 5 may require close coordination between genomic scientists and breeders to obtain gametes, fertilised eggs, or larval stages required for genome editing approaches, as well as engagement with policy experts on conditions governing gene editing approval. Therefore, the availability of both a skilled workforce, and access to a sufficiently large sampling population (and the capacity to maintain the population) needs to be addressed before any further action is taken. In these considerations, it is assumed that genetic sample processing occurs externally (see section 4.0 Resources) since it is unusual for SME or conservation aquaculture programs to have their own genetic labouratory capacity.
Once costs and practical considerations have been addressed, it is also important to understand the limitations of different genomic approaches. Individual DNA-based identification using DNA barcoding requires a set of informative markers, but the marker informativeness depends on polymorphism rates, a parameter itself dependent on population inbreeding and actual sample size. The information content of markers used for individual identification is therefore not stable across either time or space, requiring regular testing against a known standard to assess the usefulness of the markers used. Small marker panels can capture a high percentage of the genetic variance if the markers have been preselected (Vallejo et al., 2018), but only if they have been preselected based on previous, genome-wide analyses, noting that selected marker informativeness declines with selection (which brings these markers to fixation). Imputation partially mitigates these constraints, because it requires the genotyping of the parents, or the availability of a reference panel consisting of many individuals with a known genetic relationship between the panels and the samples that will undergo imputation (Marchini and Howie, 2010; Verbyla et al., 2022).
GWAS can provide an understanding of the genetic architecture of a trait, but sample size and marker density must be adequate to identify loci associated with a phenotype. If the marker set is too sparse, or if the sample size is too small, power might not be sufficient to identify loci that are associated with the phenotype. It is also important to remember that GWAS identify genomic locations associated with a phenotype, generally covering many loci, and that the association may be indirect between a marker locus and trait, and thus any locus identified in a GWAS cannot be assumed causal for a phenotype. Further steps, such as fine mapping, would likely be required to identify genetic variant(s) responsible for a trait, assuming one exists (Schaid et al., 2018). Independent GWAS, conducted on the same trait(s) but on different populations can also help to validate the effect of a locus on a phenotype, with associated additional costs to support these experiments.
4 Resources for implementing genetic/genomic work
SMEs and conservation programmes interested in the adoption of genomic technologies may find support through contact and/or partnership with aquaculture societies and organisations, research consortia, governmental institutes and academic institutions, and fee-for-service providers. Support from these partners or partnerships range from advice, to access, to development and implementation of genetic and genomic tools and information, to funding dedicated to breeding or conservation. For instance, in Canada, the Fundy Salmon Recovery program is supported in part by aquaculture companies and via an aquaculture association (https://www.canada.ca/en/parks-canada/news/2017/10/fundy_salmon_recovery.html).
Aquaculture societies, both national and international, (for example in the ICES region, the European Aquaculture Society (https://aquaeas.eu); the World Aquaculture Society (https://www.was.org); the U.S. Aquaculture Society (https://www.usaquaculture.org/); the American Fisheries Society (https://fishculture.fisheries.org)), can provide specific know-how, recommendations on service providers, and information about available funding. While the focus of aquaculture societies might be primarily business related, the importance of sustainability and conservation in the aquaculture sector means that aquaculture societies may support collabouration or supporting conservation programs, especially those focused on the wild counterparts of farmed species.
Another source of support for SME and conservation programs are research consortia. Research consortia might be permanent bodies akin to aquaculture societies, organisations and projects, or might be limited in time and scope, and specifically created to apply for domestic or international funding opportunities. Drivers of the latter type of consortia is the availability of national or international funding, such as the EU Horizon funding instruments, which require the involvement of industrial/non-academic partners in consortia applying for funding. Active involvement in research or trade consortia could allow SMEs and conservation programmes to increase their visibility and their professional network, facilitating their inclusion in consortia responding to funding calls.
Academic, institutional research organisations or institutes, government agencies, and intergovernmental organisations often have a sustained interest in aquaculture and/or conservation breeding, and thus a partnership with these research institutions can facilitate access to the pertinent know-how concerning the acquisition, use, and analysis of genomic data. Depending on funding, research institutes can provide access to specific infrastructure, and grant agreements may cover specific costs (such as genotyping, personnel, data analysis), decreasing the investment required by the SME. It is important to remember that, unless working as service providers, research institutes and funding agencies expect dissemination of results, the details of which should be agreed upon before work commences.
In addition to these resources, fee-for-service providers are also available to help guide, develop, and implement the use of genomic approaches within the breeding programs. There are several that have extensive experience in, or are specifically focused on, the aquaculture sector, and many of these companies have worked on a broad array of species. Because providers may vary by region or change rapidly within this growing field, specific providers are not listed here. However, many of the above resources would be able to provide information on service providers working in a region or on a specific species. If financial resources permit, progress is likely to be the quickest through this route. This investment may be particularly worthwhile for emerging aquaculture species, which may not yet attract attention from major funding bodies. This can lead to challenges in acquiring funding and developing institutional or academic collabourations, as well as in achieving the critical mass required to establish research consortia focused on that species. It should be noted, however, that there may be funding opportunities specifically geared towards development of newer species for both commercial and conservation programs.
5 Summary
Ultimately, the argument for the use of genetic and genomic tools and approaches by SME was stated succinctly by Gjedrem et al. (2012): “investments in well planned and managed breeding programs are unique, because genetic gains obtained in such programs are eternal and cumulative”.
Significant genetic gains are possible and can be achieved more rapidly through the use of DNA-informed breeding, which is broadly defined to include any DNA-based information, from low-coverage markers through genomic selection and genome-wide selection, to inform breeding decisions (Peace, 2017).
Long-term conservation of threatened aquatic species cannot be achieved without considering genetic diversity and proper broodstock management (Kozfkay et al., 2008). In some cases, with extremely threatened species, what is lost from genetic diversity in captive supportive breeding due to inadequate measures, is lost forever (Frankham, 2010; Bossu et al., 2023).
Acknowledgments
The authors would like to thank the ICES Working Group on the Application of Genetics for Fisheries and Aquaculture for initiating this collabouration. We thank Nick Jeffery and Steve Leadbeater from the Department of Fisheries and Oceans (DFO), and John Hyde (NOAA Fisheries) for their valuable feedback on an earlier version of this manuscript. We also thank Rolf B. Edvardsen from the Institute of Marine Research for his valuable feedback on specific sections of this manuscript. The manuscript benefited from the insightful comments and suggestions of three anonymous reviewers. Together, their expertise and constructive criticism were instrumental in enhancing the quality of this work.
Data availability statement
Data sharing is not applicable to this article as no new data were created or analyzed in this review.
References
- Abdelrahman H, ElHady M, Alcivar-Warren A, Allen S, Al-Tobasei R, Bao LS, Beck B, Blackburn H, Bosworth B, Buchanan J, Chappell J, Daniels W, Dong S, Dunham R, Durland E, Elaswad A, Gomez-Chiarri M, Gosh K, Guo XM, Hackett P, Hanson T, Hedgecock D, Howard T, Holland L, Jackson M, Jin YL, Khalil K, Kocher T, Leeds T, Li N, Lindsey L, Liu SK, Liu ZJ, Martin K, Novriadi R, Odin R, Palti Y, Peatman E, Proestou D, Qin GY, Reading B, Rexroad C, Roberts S, Salem M, Severin A, Shi HT, Shoemaker C, Stiles S, Tan SX, Tang KFJ, Thongda W, Tiersch T, Tomasso J, Prabowo WT, Vallejo R, van der Steen F H, Vo K, Waldbieser G, Wang HP, Wang XZ, Xiang JH, Yang YJ, Yant R, Yuan ZH, Zeng QF, Zhou T, Bre AGG. 2017. Aquaculture genomics, genetics and breeding in the United States: current status, challenges, and priorities for future research. BMC Genomics 18: 235. [CrossRef] [PubMed] [Google Scholar]
- Abdollahi-Arpanahi LR, Gianola D, Peñagaricano F. 2020. Deep learning versus parametric and ensemble methods for genomic prediction of complex phenotypes. Genet Sel Evol 52: 12. [CrossRef] [PubMed] [Google Scholar]
- Anderson JH, Pess GR, Carmichael RW, Ford MJ, Cooney TD, Baldwin CM, McClure MM. 2014. Planning Pacific salmon and steelhead reintroductions aimed at long-term viability and recovery. N Am J Fish Manage 34: 72–93. [CrossRef] [Google Scholar]
- Araki H, Cooper B, Blouin MS. 2007. Genetic effects of captive breeding cause a rapid, cumulative fitness decline in the wild. Science 318: 100–103. [CrossRef] [PubMed] [Google Scholar]
- Ashton DT, Hilario E, Jaksons P, Ritchie PA, Wellenreuther M. 2019. Genetic diversity and heritability of economically important traits in captive Australasian snapper (Chrysophrys auratus). Aquaculture 505: 190–198. [CrossRef] [Google Scholar]
- Attard CRM, Möller LM, Sasaki M, Hammer MP, Bice CM, Brauer CJ, Carvalho DC, Harris JO, Beheregaray LB. 2016. A novel holistic framework for genetic-based captive-breeding and reintroduction programs. Conserv Biol 30: 1060–1069. [CrossRef] [PubMed] [Google Scholar]
- Auld HL, Jacobson DP, Rhodes AC, Banks MA. 2021. Differences in mate pairings of hatchery- and natural-origin Coho salmon inferred from offspring genotypes. Integr Organism Biol 3: obab020. [CrossRef] [Google Scholar]
- Azodi CB, Bolger E, McCarren A, Roantree M, de los Campos G, Shiu SH. 2019. Benchmarking parametric and machine learning models for genomic prediction of complex traits. G3-Genes Genom Genet 9: 3691–3702. [Google Scholar]
- Baetscher DS, Clemento AJ, Ng TC, Anderson EC, Garza JC. 2018. Microhaplotypes provide increased power from short-read DNA sequences for relationship inference. Mol Ecol Resour 18: 296–305. [CrossRef] [PubMed] [Google Scholar]
- Balon EK. 2004. About the oldest domesticates among fishes. J Fish Biol 65(s1): 1–27. [Google Scholar]
- Barria A, Benzie JAH, Houston RD, De Koning DJ, de Verdal H. 2021. Genomic selection and genome-wide association study for feed-efficiency traits in a farmed Nile tilapia (Oreochromis niloticus) population. Front Genet 12: 737906. [CrossRef] [PubMed] [Google Scholar]
- Barson NJ, Aykanat T, Hindar K, Baranski M, Bolstad GH, Fiske P, Jacq C, Jensen AJ, Johnston SE, Karlsson S, Kent M, Oen TM, Niemelä E, Nome T, Næsje TF, Orell P, Romakkaniemi A, Sægrov H, Urdal K, Erkinaro J, Lien S, Primmer CR. 2015. Sex-dependent dominance at a single locus maintains variation in age at maturity in salmon. Nature 528: 405–408. [CrossRef] [PubMed] [Google Scholar]
- Bay RA, Harrigan RJ, Underwood VL, Gibbs HL, Smith TB, Ruegg K. 2018. Genomic signals of selection predict climate-driven population declines in a migratory bird. Science 359: 83–86. [CrossRef] [PubMed] [Google Scholar]
- Bean TP, Tanguy A, Peñaloza C, Gundappa MK, Boutet I, Houston RD, Macqueen DJ, Boudry P. 2022. Two parallel chromosome-level reference genomes to support restoration and aquaculture of European flat oyster Ostrea edulis. Evol Appl 15: 1709–1712. [CrossRef] [PubMed] [Google Scholar]
- Bernatchez S, Xuereb A, Laporte M, Benestan L, Steeves R, Laflamme M, Bernatchez L, Mallet MA. 2019. Seascape genomics of eastern oyster (Crassostrea virginica) along the Atlantic coast of Canada. Evol Appl 12: 587–609. [CrossRef] [PubMed] [Google Scholar]
- Besnier F, Ayllon F, Skaala O, Solberg MF, Fjeldheim PT, Anderson K, Knutar S, Glover KA. 2022. Introgression of domesticated salmon changes life history and phenology of a wild salmon population. Evol Appl 15: 853–864. [CrossRef] [PubMed] [Google Scholar]
- Besson M, Allal F, Chatain B, Vergnet A, Clota F, Vandeputte M. 2019. Combining individual phenotypes of feed intake with genomic data to improve feed efficiency in sea bass. Front Genet 10: 219. [CrossRef] [PubMed] [Google Scholar]
- Besson M, Rombout N, Salou G, Vergnet A, Cariou S, Bruant JS, Izquierdo M, Bestin A, Clota F, Haffray P, Allal F, Vandeputte M. 2022. Potential for genomic selection on feed efficiency in gilthead sea bream (Sparus aurata), based on individual feed conversion ratio, carcass and lipid traits. Aquacult Rep 24: 101171. [Google Scholar]
- Bolstad GH, Hindar K, Robertsen G, Jonsson B, Sægrov H, Diserud OH, Fiske P, Jensen AJ, Urdal K, Næsje TF, Barlaup BT, Floro-Larsen B, Lo H, Niemelä E, Karlsson S. 2017. Gene flow from domesticated escapes alters the life history of wild Atlantic salmon. Nat Ecol Evol 1: 124. [CrossRef] [PubMed] [Google Scholar]
- Bolstad GH, Karlsson S, Hagen IJ, Fiske P, Urdal K, Sægrov H, Floro-Larsen B, Sollien VP, Ostborg G, Diserud OH, Jensen AJ, Hindar K. 2021. Introgression from farmed escapees affects the full life cycle of wild Atlantic salmon. Sci Adv 7: eabj3397. [CrossRef] [PubMed] [Google Scholar]
- Bootsma ML, Gruenthal KM, McKinney GJ, Simmons L, Miller L, Sass GG, Larson WA. 2020. A GT-seq panel for walleye (Sander vitreus) provides important insights for efficient development and implementation of amplicon panels in non-model organisms. Mol Ecol Resour 20: 1706–1722. [CrossRef] [PubMed] [Google Scholar]
- Bossu CM, Rodriguez M, Rayne C, Chromczak DA, Higgins PG, Trulio LA, Ruegg KC. 2023. Genomic approaches to mitigating genetic diversity loss in declining populations. Mol Ecol 32: 5228–5240. [CrossRef] [PubMed] [Google Scholar]
- Boudry P, Allal F, Aslam ML, Bargelloni L, Bean TP, Brard-Fudulea S, Brieuc MSO, Calboli FCF, Gilbey J, Haffray P, Lamy JB, Morvezen R, Purcell C, Prodohl PA, Vandeputte M, Waldbieser GC, Sonesson AK, Houston RD. 2021. Current status and potential of genomic selection to improve selective breeding in the main aquaculture species of International Council for the Exploration of the Sea (ICES) member countries. Aquacult Rep 20: 100700. [Google Scholar]
- Boutet I, Monteiro HJA, Baudry L, Takeuchi T, Bonnivard E, Billoud B, Farhat S, Gonzales-Araya R, Salaun B, Andersen AC, Toullec JY, Lallier FH, Flot JF, Guiglielmoni N, Guo XM, Li C, Allam B, Pales-Espinosa E, Hemmer-Hansen J, Moreau P, Marbouty M, Koszul R, Tanguy A. 2022. Chromosomal assembly of the flat oyster (Ostrea edulis L.) genome as a new genetic resource for aquaculture. Evol Appl 15: 1730–1748. [CrossRef] [PubMed] [Google Scholar]
- Bradbury IR, Lehnert SJ, Kess T, Van Wyngaarden M, Duffy S, Messmer AM, Wringe B, Karoliussen S, Dempson JB, Fleming IA, Solberg MF, Glover KA, Bentzen P. 2022. Genomic evidence of recent European introgression into North American farmed and wild Atlantic salmon. Evol Appl 15: 1436–1448. [CrossRef] [PubMed] [Google Scholar]
- Bradbury IR, Wringe BF, Watson B, Paterson I, Horne J, Beiko R, Lehnert SJ, Clément M, Anderson EC, Jeffery NW, Duffy S, Sylvester E, Robertson M, Bentzen P. 2018. Genotyping-by-sequencing of genome-wide microsatellite loci reveals fine-scale harvest composition in a coastal Atlantic salmon fishery. Evol Appl 11: 918–930. [CrossRef] [PubMed] [Google Scholar]
- Brieuc MSO, Waters CD, Drinan DP, Naish KA. 2018. A practical introduction to random forest for genetic association studies in ecology and evolution. Mol Ecol Resour 18: 755–766. [CrossRef] [PubMed] [Google Scholar]
- Capblancq T, Fitzpatrick MC, Bay RA, Exposito-Alonso M, Keller SR. 2020. Genomic prediction of (mal)adaptation across current and future climatic landscapes. Annu Rev Ecol Evol Syst 51: 245–269. [CrossRef] [Google Scholar]
- Casey J, Jardim E, Martinsohn JT. 2016. The role of genetics in fisheries management under the EU common fisheries policy. J Fish Biol 89: 2755–2767. [CrossRef] [PubMed] [Google Scholar]
- Catanach A, Ruigrok M, Bowatte D, Davy M, Storey R, Valenza-Troubat N, López-Girona E, Hilario E, Wylie MJ, Chagné D, Wellenreuther M. 2021. The genome of New Zealand trevally (Carangidae: Pseudocaranx georgianus) uncovers a XY sex determination locus. BMC Genomics 22: 302. [CrossRef] [PubMed] [Google Scholar]
- Chang SL, Ward HGM, Russello MA. 2021. Genotyping-in-thousands by sequencing panel development and application to inform kokanee salmon (Oncorhynchus nerka) fisheries management at multiple scales. PLoS One 16: e0261966. [CrossRef] [PubMed] [Google Scholar]
- Chen Z. 2020. Proof of concept: efficacy of cleaner fish, cultured juvenile cunner (Tautogolabrus adsperus), for sea lice (Lepeophtheirus salmonis) mitigation and control in Atlantic salmon (Salmo salar). PhD thesis, Memorial University of Newfoundland. Available at: http://research.library.mun.ca/id/eprint/15071 [Google Scholar]
- Chistiakov DA, Hellemans B, Volckaert FAM. 2006. Microsatellites and their genomic distribution, evolution, function and applications: a review with special reference to fish genetics. Aquaculture 255: 1–29. [CrossRef] [Google Scholar]
- Christie MR, French RA, Marine ML, Blouin MS. 2013. How much does inbreeding contribute to the reduced fitness of hatchery-born steelhead (Oncorhynchus mykiss) in the wild? J Hered 105: 111–119. [Google Scholar]
- Christie MR, Marine ML, French RA, Waples RS, Blouin MS. 2012. Effective size of a wild salmonid population is greatly reduced by hatchery supplementation. Heredity 109: 254–260. [CrossRef] [PubMed] [Google Scholar]
- Colihueque N. 2010. Genetics of salmonid skin pigmentation: clues and prospects for improving the external appearance of farmed salmonids. Rev Fish Biol Fish 20: 71–86. [CrossRef] [Google Scholar]
- Colihueque N, Araneda C. 2014. Appearance traits in fish farming: progress from classical genetics to genomics, providing insight into current and potential genetic improvement. Front Genet 5: 251. [CrossRef] [PubMed] [Google Scholar]
- Colsoul B, Boudry P, Pérez-Parallé ML, Bratoš Cetinić A, Hugh-Jones T, Arzul I, Mérou N, Wegner KM, Peter C, Merk V, Pogoda B. 2021. Sustainable large-scale production of European flat oyster (Ostrea edulis) seed for ecological restoration and aquaculture: a review. Rev Aquacult 13: 1423–1468. [CrossRef] [Google Scholar]
- Costa I, Hamoutene D, Murray H, Lush L, Burt K, Eaves A, Keng P. 2016. Documentation of cunner (Tautogolabrus adspersus) cleaning behaviour in tanks with Atlantic salmon (Salmo salar) smolts infested with sea lice (Lepeophtheirus salmonis). Can Tech Rep Fish Aquat Sci 3168: iv + 11 p. [Google Scholar]
- D'Agaro E, Favaro A, Matiussi S, Gibertoni PP, Esposito S. 2021. Genomic selection in salmonids: new discoveries and future perspectives. Aquacult Int 29: 2259–2289. [CrossRef] [Google Scholar]
- D'Ambrosio J, Phocas F, Haffray P, Bestin A, Brard-Fudulea S, Poncet C, Quillet E, Dechamp N, Fraslin C, Charles M, Dupont-Nivet M. 2019. Genome-wide estimates of genetic diversity, inbreeding and effective size of experimental and commercial rainbow trout lines undergoing selective breeding. Genet Sel Evol 51: 26. [CrossRef] [PubMed] [Google Scholar]
- Davidson WS, Birt TP, Green JM. 1989. A review of genetic variation in Atlantic salmon, Salmo salar L., and its importance for stock identification, enhancement programs and aquaculture. J Fish Biol 34: 547–560. [CrossRef] [Google Scholar]
- Devlin RH, Sundström LF, Leggatt RA. 2015. Assessing ecological and evolutionary consequences of growth-accelerated genetically engineered fishes. Bioscience 65: 685–700. [CrossRef] [Google Scholar]
- Devlin RH, Sundström LF, Muir WM. 2006. Interface of biotechnology and ecology for environmental risk assessments of transgenic fish. Trends Biotechnol 24: 89–97. [CrossRef] [PubMed] [Google Scholar]
- DFO. 2018. Environmental and indirect human health risk assessment of the Glofish® Electric Green® Tetra and the Glofish® Long-Fin Electric Green® Tetra (Gymnocorymbus ternetzi): a transgenic ornamental fish. DFO Can. Sci. Advis. Sec. Sci. Advis. Rep. 2018/027. [Google Scholar]
- DFO. 2018. Review of the science associated with the Inner Bay of Fundy Atlantic salmon Live Gene Bank and supplementation programs. DFO Can. Sci. Advis. Sec. Sci. Advis. Rep. 2018/041. [Google Scholar]
- DFO. 2019. Environmental and indirect human health risk assessment of the GloFish® Tetras (Gymnocorymbus ternetzi): five lines of transgenic ornamental fish. DFO Can. Sci. Advis. Sec. Sci. Advis. Rep. 2019/002. [Google Scholar]
- DFO. 2020. Environmental and indirect human health risk assessment of the GloFish® Cosmic Blue® and Galactic Purple® Danios (Danio rerio): transgenic ornamental fish. DFO Can. Sci. Advis. Sec. Sci. Advis. Rep. 2020/016. [Google Scholar]
- DFO. 2020. Environmental and indirect human health risk assessment of the GloFish® Sunburst Orange® Danio (Danio rerio): a transgenic ornamental fish. DFO Can. Sci. Advis. Sec. Sci. Advis. Rep. 2020/015. [Google Scholar]
- DFO. 2021. Environmental and indirect human health risk assessment of the GloFish® Bettas: three lines of transgenic ornamental fish. DFO Can. Sci. Advis. Sec. Sci. Advis. Rep. 2021/046. [Google Scholar]
- Diamond J. 2002. Evolution, consequences and future of plant and animal domestication. Nature 418: 700–707. [CrossRef] [PubMed] [Google Scholar]
- Dietrich C, Leggatt R, McGowan C. 2022. Environmental risk assessment of the GloFish® Starfire Red®, Electric Green®, Sunburst Orange®, and Galactic Purple® Barbs (Puntigrus tetrazona): transgenic ornamental fishes. DFO Can. Sci. Advis. Sec. Res. Doc. 2022/077: vii + 34 p. [Google Scholar]
- Divilov K, Merz N, Schoolfield B, Green TJ, Langdon C. 2023. Marker-assisted selection in a Pacific oyster population for an antiviral QTL conferring increased survival to OsHV-1 mortality events in Tomales Bay. Aquaculture 567: 739217. [Google Scholar]
- Doudna JA, Charpentier E. 2014. The new frontier of genome engineering with CRISPR-Cas9. Science 346: 1258096. [Google Scholar]
- Du SJ, Gong ZY, Fletcher GL, Shears MA, King MJ, Idler DR, Hew CL. 1992. Growth enhancement in transgenic Atlantic salmon by the use of an all fish chimeric growth-hormone gene construct. Bio-Technol 10: 176–181. [Google Scholar]
- Einfeldt AL, Kess T, Messmer A, Duffy S, Wringe BF, Fisher J, den Heyer C, Bradbury IR, Ruzzante DE, Bentzen P. 2021. Chromosome level reference of Atlantic halibut Hippoglossus hippoglossus provides insight into the evolution of sexual determination systems. Mol Ecol Resour 21: 1686–1696. [CrossRef] [PubMed] [Google Scholar]
- Enbody ED, Pettersson ME, Sprehn CG, Palm S, Wickström H, Andersson L. 2021. Ecological adaptation in European eels is based on phenotypic plasticity. Proc Natl Acad Sci USA 118: e2022620118. [CrossRef] [PubMed] [Google Scholar]
- Espiñeira M, Vieites JM. 2016. Genetic system for an integral traceability of European eel (Anguilla anguilla) in aquaculture and seafood products: authentication by fast real-time PCR. Eur Food Res Technol 242: 25–31. [CrossRef] [Google Scholar]
- Euclide PT, Larson WA, Bootsma M, Miller LM, Scribner KT, Stott W, Wilson CC, Latch EK. 2022. A new GTSeq resource to facilitate multijurisdictional research and management of walleye Sander vitreus. Ecol Evol 12: e9591. [CrossRef] [PubMed] [Google Scholar]
- Fernández Sánchez JL, Llorente I, Basurco B, Aguilera C. 2022. Assessing the economic impact of key operational factors on grow-out farms producing European sea bass under different scenarios of production. Aquacult Econ Manag 26: 232–250. [CrossRef] [Google Scholar]
- Frankham R. 2010. Challenges and opportunities of genetic approaches to biological conservation. Biol Conserv 143: 1919–1927. [CrossRef] [Google Scholar]
- Fuji K, Hasegawa O, Honda K, Kumasaka K, Sakamoto T, Okamoto N. 2007. Marker-assisted breeding of a lymphocystis disease-resistant Japanese flounder (Paralichthys olivaceus). Aquaculture 272: 291–295. [CrossRef] [Google Scholar]
- Gaj T, Gersbach CA, Barbas CF. 2013. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol 31: 397–405. [CrossRef] [PubMed] [Google Scholar]
- Gao GT, Waldbieser GC, Youngblood RC, Zhao DY, Pietrak MR, Allen MS, Stannard JA, Buchanan JT, Long RL, Milligan M, Burr G, Mejia-Guerra K, Sheehan MJ, Scheffler BE, Rexroad CE, Peterson BC, Palti Y. 2023. The generation of the first chromosome-level de novo genome assembly and the development and validation of a 50K SNP array for the St. John River aquaculture strain of North American Atlantic salmon. G3-Genes Genom Genet 13. [Google Scholar]
- Garcia ALS, Bosworth B, Waldbieser G, Misztal I, Tsuruta S, Lourenco DAL. 2018. Development of genomic predictions for harvest and carcass weight in channel catfish. Genet Sel Evol 50: 66. [CrossRef] [PubMed] [Google Scholar]
- Gautason E, Sahana G, Guldbrandtsen B, Berg P. 2022. Optimum contribution selection in a dairy cattle population with different relationship matrices. In: Proc 12th World Congress on Genetics Applied to Livestock Production (WCGALP), Wageningen Academic Publishers, 2769–2772. [CrossRef] [Google Scholar]
- Gebregiwergis GT, Sorensen AC, Henryon M, Meuwissen T. 2020. Controlling coancestry and thereby future inbreeding by optimum-contribution selection using alternative genomic-relationship matrices. Front Genet 11: 345. [CrossRef] [PubMed] [Google Scholar]
- Ghildiyal K, Nayak SS, Rajawat D, Sharma A, Chhotaray S, Bhushan B, Dutt T, Panigrahi M. 2023. Genomic insights into the conservation of wild and domestic animal diversity: a review. Gene 886: 147719. [CrossRef] [PubMed] [Google Scholar]
- Ginson R, Walter RP, Mandrak NE, Beneteau CL, Heath DD. 2015. Hierarchical analysis of genetic structure in the habitat-specialist Eastern Sand Darter (Ammocrypta pellucida). Ecol Evol 5: 695–708. [CrossRef] [PubMed] [Google Scholar]
- Gjedrem T. 1985. Improvement of productivity through breeding schemes. GeoJournal 10: 233–241. [CrossRef] [Google Scholar]
- Gjedrem T, Gjoen HM, Gjerde B. 1991. Genetic origin of Norwegian farmed Atlantic salmon. Aquaculture 98: 41–50. [CrossRef] [Google Scholar]
- Gjedrem T, Robinson N, Rye M. 2012. The importance of selective breeding in aquaculture to meet future demands for animal protein: a review. Aquaculture 350: 117–129. [CrossRef] [Google Scholar]
- Gong J, Zhao J, Ke QZ, Li BJ, Zhou ZX, Wang JY, Zhou T, Zheng WQ, Xu P. 2022. First genomic prediction and genome-wide association for complex growth-related traits in Rock Bream (Oplegnathus fasciatus). Evol Appl 15: 523–536. [CrossRef] [PubMed] [Google Scholar]
- Gundappa MK, Peñaloza C, Regan T, Boutet I, Tanguy A, Houston RD, Bean TP, Macqueen DJ. 2022. Chromosome-level reference genome for European flat oyster (Ostrea edulis L.). Evol Appl 15: 1713–1729. [CrossRef] [PubMed] [Google Scholar]
- Gundappa MK, Robledo D, Hamilton A, Houston RD, Prendergast JGD, Macqueen DJ. 2023. High performance imputation of structural and single nucleotide variants in Atlantic salmon using low-coverage whole genome sequencing. bioRxiv, https://doi.org/10.1101/2023.03.05.531147. [Google Scholar]
- Güralp H, Skaftnesmo KO, Kjærner-Semb E, Straume AH, Kleppe L, Schulz RW, Edvardsen RB, Wargelius A. 2020. Rescue of germ cells in dnd crispant embryos opens the possibility to produce inherited sterility in Atlantic salmon. Sci Rep-UK 10: 18042. [CrossRef] [Google Scholar]
- Gutierrez AP, Matika O, Bean TP, Houston RD. 2018. Genomic selection for growth traits in Pacific oyster (Crassostrea gigas): potential of low-density marker panels for breeding value prediction. Front Genet 9: 391. [CrossRef] [PubMed] [Google Scholar]
- Gutierrez AP, Turner F, Gharbi K, Talbot R, Lowe NR, Peñaloza C, McCullough M, Prodöhl PA, Bean TP, Houston RD. 2017. Development of a medium density combined-species SNP array for Pacific and European oysters (Crassostrea gigas and Ostrea edulis). G3-Genes Genom Genet 7: 2209–2218. [CrossRef] [Google Scholar]
- Hagen IJ, Jensen AJ, Bolstad GH, Diserud OH, Hindar K, Lo H, Karlsson S. 2019. Supplementary stocking selects for domesticated genotypes. Nat Commun 10: 199. [CrossRef] [PubMed] [Google Scholar]
- Hagen IJ, Ugedal O, Jensen AJ, Lo H, Holthe E, Bjoru B, Floro-Larsen B, Sægrov H, Skoglund H, Karlsson S. 2021. Evaluation of genetic effects on wild salmon populations from stock enhancement. ICES J Mar Sci 78: 900–909. [CrossRef] [Google Scholar]
- Håstein T, Hill BJ, Berthe F, Lightner DV. 2001. Traceability of aquatic animals. Rev Sci Tech OIE 20: 564–583. [CrossRef] [PubMed] [Google Scholar]
- Herlin M, Delghandi M, Wesmajervi M, Taggart JB, McAndrew BJ, Penman DJ. 2008. Analysis of the parental contribution to a group of fry from a single day of spawning from a commercial Atlantic cod (Gadus morhua) breeding tank. Aquaculture 274: 218–224. [CrossRef] [Google Scholar]
- Herlin M, Taggart JB, McAndrew BJ, Penman DJ. 2007. Parentage allocation in a complex situation: a large commercial Atlantic cod (Gadus morhua) mass spawning tank. Aquaculture 272 (Suppl. 1): S195– S203. [CrossRef] [Google Scholar]
- Hess JE, Zendt JS, Matala AR, Narum SR. 2016. Genetic basis of adult migration timing in anadromous steelhead discovered through multivariate association testing. Proc R Soc B-Biol Sci 283: 20153064. [CrossRef] [PubMed] [Google Scholar]
- Hoban S, Kelley JL, Lotterhos KE, Antolin MF, Bradburd G, Lowry DB, Poss ML, Reed LK, Storfer A, Whitlock MC. 2016. Finding the genomic basis of local adaptation: pitfalls, practical solutions, and future directions. Am Nat 188: 379–397. [CrossRef] [PubMed] [Google Scholar]
- Holborn MK, Ang KP, Elliott JAK, Powell F, Boulding EG. 2020. Genome wide analysis of infectious salmon anemia resistance in commercial Saint John River Atlantic salmon. Aquaculture 514. [Google Scholar]
- Holborn MK, Einfeldt AL, Kess T, Duffy SJ, Messmer AM, Langille BL, Brachmann MK, Gauthier J, Bentzen P, Knutsen TM, Kent M, Boyce D, Bradbury IR. 2022. Reference genome of lumpfish Cyclopterus lumpus Linnaeus provides evidence of male heterogametic sex determination through the AMH pathway. Mol Ecol Resour 22: 1427–1439. [CrossRef] [PubMed] [Google Scholar]
- Holborn MK, Kess T, Nugent CM, Brodeur NN, Adesola J, Cronmiller E, Hamilton LC, Jones RA, Lenentine BL, Macdonnell A, Mcbride M, Messmer A, de Mestral L, Moreau DTR, Wilson T, Bradbury IR, Wringe BF. 2025. Improved estimation of aquaculture associated European introgression in a captive breeding program for endangered Atlantic salmon. Conserv Genet 26: 235–247. [Google Scholar]
- Holborn MK, Rochus CM, Ang KP, Elliott JAK, Leadbeater S, Powell F, Boulding EG. 2019. Family-based genome wide association analysis for salmon lice (Lepeophtheirus salmonis) resistance in North American Atlantic salmon using a 50K SNP array. Aquaculture 511: 734215. [CrossRef] [Google Scholar]
- Holman LE, de la Serrana DG, Onoufriou A, Hillestad B, Johnston IA. 2017. A workflow used to design low density SNP panels for parentage assignment and traceability in aquaculture species and its validation in Atlantic salmon. Aquaculture 476: 59–64. [CrossRef] [Google Scholar]
- Homburger JR, Neben CL, Mishne G, Zhou AY, Kathiresan S, Khera AV. 2019. Low coverage whole genome sequencing enables accurate assessment of common variants and calculation of genome-wide polygenic scores. Genome Med 11: 74. [CrossRef] [PubMed] [Google Scholar]
- Horn RL, Hess M, Harmon S, Hess J, Delomas TA, Campbell MR, Narum S. 2023. Multigeneration pedigrees to monitor hatchery broodstock composition and genetic variation of spring/summer Chinook salmon in the Columbia River Basin. N Am J Fish Manage 43: 794–820. [CrossRef] [Google Scholar]
- Horn RL, Kamphaus C, Murdoch K, Narum SR. 2020. Detecting genomic variation underlying phenotypic characteristics of reintroduced Coho salmon (Oncorhynchus kisutch). Conserv Genet 21: 1011–1021. [CrossRef] [Google Scholar]
- Horreo JL, Machado-Schiaffino G, Griffiths A, Bright D, Stevens J, Garcia-Vazquez E. 2008. Identification of differential broodstock contribution affecting genetic variability in hatchery stocks of Atlantic salmon (Salmo salar). Aquaculture 280: 89–93. [CrossRef] [Google Scholar]
- Houston RD, Bean TP, Macqueen DJ, Gundappa MK, Jin YH, Jenkins TL, Selly SLC, Martin SAM, Stevens JR, Santos EM, Davie A, Robledo D. 2020. Harnessing genomics to fast-track genetic improvement in aquaculture. Nat Rev Genet 21: 389–409. [CrossRef] [PubMed] [Google Scholar]
- Houston RD, Davey JW, Bishop SC, Lowe NR, Mota-Velasco JC, Hamilton A, Guy DR, Tinch AE, Thomson ML, Blaxter ML, Gharbi K, Bron JE, Taggart JB. 2012. Characterisation of QTL-linked and genome-wide restriction site-associated DNA (RAD) markers in farmed Atlantic salmon. BMC Genomics 13: 244. [CrossRef] [PubMed] [Google Scholar]
- Houston RD, Haley CS, Hamilton A, Guy DR, Mota-Velasco JC, Gheyas AA, Tinch AE, Taggart JB, Bron JE, Starkey WG, McAndrew BJ, Verner-Jeffreys DW, Paley RK, Rimmer GSE, Tew IJ, Bishop SC. 2010. The susceptibility of Atlantic salmon fry to freshwater infectious pancreatic necrosis is largely explained by a major QTL. Heredity 105: 318–327. [CrossRef] [PubMed] [Google Scholar]
- Hurt C, Harman A. 2017. Development of polymorphic microsatellite loci for the design of management and conservation strategies of the critically endangered Barrens topminnow (Fundulus julisia, Williams & Etnier, 1982). J Appl Ichthyol 33: 797–800. [CrossRef] [Google Scholar]
- Jackson TR, Martin-Robichaud DJ, Reith ME. 2003. Application of DNA markers to the management of Atlantic halibut (Hippoglossus hippoglossus) broodstock. Aquaculture 220: 245–259. [CrossRef] [Google Scholar]
- Janssen K, Saatkamp H, Komen H. 2018. Cost-benefit analysis of aquaculture breeding programs. Genet Sel Evol 50: 2. [CrossRef] [PubMed] [Google Scholar]
- Jansson E, Faust E, Bekkevold D, Quintela M, Durif C, Halvorsen KT, Dahle G, Pampoulie C, Kennedy J, Whittaker B, Unneland L, Post S, André C, Glover KA. 2023. Global, regional, and cryptic population structure in a high gene-flow transatlantic fish. PLOS One 18: e0283351. [CrossRef] [PubMed] [Google Scholar]
- Jeffery NW, Vercaemer B, Stanley RRE, Kess T, Dufresne F, Noisette F, O'Connor MI, Wong MC. 2024. Variation in genomic vulnerability to climate change across temperate populations of eelgrass (Zostera marina). Evol Appl 17: e13671. [CrossRef] [PubMed] [Google Scholar]
- Jeffery NW, Wringe BF, McBride MC, Hamilton LC, Stanley RRE, Bernatchez L, Kent M, Clément M, Gilbey J, Sheehan TF, Bentzen P, Bradbury IR. 2018. Range-wide regional assignment of Atlantic salmon (Salmo salar) using genome-wide single-nucleotide polymorphisms. Fish Res 206: 163–175. [CrossRef] [Google Scholar]
- Jerry DR, Jones DB, Lillehammer M, Massault C, Loughnan S, Cate HS, Harrison PJ, Strugnell JM, Zenger KR, Robinson NA. 2022. Predicted strong genetic gains from the application of genomic selection to improve growth related traits in barramundi. Aquaculture 549. [Google Scholar]
- Jones LF, Lou RN, Murray CS, Robert D, Bourne CM, Bouchard C, Kucka M, Chan YF, Carlon DB, Wiley DN, Therkildsen NO, Baumann H. 2023. Two distinct population clusters of northern sand lance (Ammodytes dubius) on the northwest Atlantic shelf revealed by whole genome sequencing. Ices J Mar Sci 80: 122–132. [CrossRef] [Google Scholar]
- Jónsdóttir ODB, Schregel J, Hagen SB, Tobiassen C, Aarnes SG, Imsland AKD. 2018. Population genetic structure of lumpfish along the Norwegian coast: aquaculture implications. Aquacult Int 26: 49–60. [CrossRef] [Google Scholar]
- Jorsboe E, Albrechtsen A. 2022. Efficient approaches for large-scale GWAS with genotype uncertainty. G3-Genes Genom Genet 12. [Google Scholar]
- Joshi R, Skaarud A, de Vera M, Alvarez AT, Ødegård J. 2020. Genomic prediction for commercial traits using univariate and multivariate approaches in Nile tilapia (Oreochromis niloticus). Aquaculture 516. [Google Scholar]
- Joung JK, Sander JD. 2013. INNOVATION TALENs: a widely applicable technology for targeted genome editing. Nat Rev Mol Cell Bio 14: 49–55. [CrossRef] [PubMed] [Google Scholar]
- Jourdan A, Morvezen R, Enez F, Haffray P, Lange A, Vétois E, Allal F, Phocas F, Bugeon J, Dégremont L, Boudry P. 2023. Potential of genomic selection for growth, meat content and colour traits in mixed-family breeding designs for the Pacific oyster Crassostrea gigas. Aquaculture 576: 739878. [CrossRef] [Google Scholar]
- Kankainen M, Setälä J, Kause A, Quinton C, Airaksinen S, Koskela J. 2016. Economic values of supply chain productivity and quality traits calculated for a farmed European whitefish breeding program. Aquacult Econ Manag 20: 131–164. [CrossRef] [Google Scholar]
- Karlsson S, Diserud OH, Fiske P, Hindar K. 2016. Widespread genetic introgression of escaped farmed Atlantic salmon in wild salmon populations. Ices J Mar Sci 73: 2488–2498. [CrossRef] [Google Scholar]
- Karlsson S, Diserud OH, Moen T, Hindar K. 2014. A standardized method for quantifying unidirectional genetic introgression. Ecol Evol 4: 3256–3263. [CrossRef] [PubMed] [Google Scholar]
- Karlsson S, Moen T, Lien S, Glover KA, Hindar K. 2011. Generic genetic differences between farmed and wild Atlantic salmon identified from a 7K SNP-chip. Mol Ecol Resour 11: 247–253. [CrossRef] [PubMed] [Google Scholar]
- Kause A, Quinton C, Airaksinen S, Ruohonen K, Koskela J. 2011. Quality and production trait genetics of farmed European whitefish, Coregonus lavaretus. J Anim Sci 89: 959–971. [CrossRef] [PubMed] [Google Scholar]
- Kause A, Ritola O, Paananen T. 2004. Breeding for improved appearance of large rainbow trout in two production environments. Aquac Res 35: 924–930. [CrossRef] [Google Scholar]
- Kause A, Ritola O, Paananen T, Mäntysaari E, Eskelinen U. 2003. Selection against early maturity in large rainbow trout Oncorhynchus mykiss: the quantitative genetics of sexual dimorphism and genotype-by-environment interactions. Aquaculture 228: 53–68. [CrossRef] [Google Scholar]
- Ke QZ, Wang JY, Bai YL, Zhao J, Gong J, Deng YC, Qu A, Suo N, Chen J, Zhou T, Xu P. 2022. GWAS and genomic prediction revealed potential for genetic improvement of large yellow croaker adapting to high plant protein diet. Aquaculture 553. [Google Scholar]
- Kess T, Bentzen P, Lehnert SJ, Sylvester EVA, Lien S, Kent MP, Sinclair-Waters M, Morris CJ, Regular P, Fairweather R, Bradbury IR. 2019. A migration-associated supergene reveals loss of biocomplexity in Atlantic cod. Sci Adv 5: eaav2461. [CrossRef] [PubMed] [Google Scholar]
- Kess T, Dempson JB, Lehnert SJ, Layton KKS, Einfeldt A, Bentzen P, Salisbury SJ, Messmer AM, Duffy S, Ruzzante DE, Nugent CM, Ferguson MM, Leong JS, Koop B, O'Connell MF, Bradbury IR. 2021. Genomic basis of deep-water adaptation in Arctic Charr (Salvelinus alpinus) morphs. Mol Ecol 30: 4415–4432. [CrossRef] [PubMed] [Google Scholar]
- Kess T, Lehnert SJ, Bentzen P, Duffy S, Messmer A, Dempson JB, Newport J, Whidden C, Robertson MJ, Chaput G, Breau C, April J, Gillis C-A., Kent M, Nugent CM, Bradbury IR. 2024. Variable parallelism in the genomic basis of age at maturity across spatial scales in Atlantic salmon. Ecol Evol 14: e11068. [CrossRef] [PubMed] [Google Scholar]
- Kijas J, Elliot N, Kube P, Evans B, Botwright N, King H, Primmer CR, Verbyla K. 2017. Diversity and linkage disequilibrium in farmed Tasmanian Atlantic salmon. Anim Genet 48: 237–241. [CrossRef] [PubMed] [Google Scholar]
- Kim SY, Lohmueller KE, Albrechtsen A, Li YR, Korneliussen T, Tian G, Grarup N, Jiang T, Andersen G, Witte D, Jorgensen T, Hansen T, Pedersen O, Wang J, Nielsen R. 2011. Estimation of allele frequency and association mapping using next-generation sequencing data. BMC Bioinformatics 12: 231. [CrossRef] [PubMed] [Google Scholar]
- Kincaid HL. 1976. Effects of inbreeding on rainbow trout populations. T Am Fish Soc 105: 273–280. [CrossRef] [Google Scholar]
- Kishimoto K, Washio Y, Yoshiura Y, Toyoda A, Ueno T, Fukuyama H, Kato K, Kinoshita M. 2018. Production of a breed of red sea bream Pagrus major with an increase of skeletal muscle mass and reduced body length by genome editing with CRISPR/Cas9. Aquaculture 495: 415–427. [CrossRef] [Google Scholar]
- Kleppe L, Fjelldal PG, Andersson E, Hansen T, Sanden M, Bruvik A, Skaftnesmo KO, Furmanek T, Kjaerner-Semb E, Crespo D, Flavell S, Pedersen AO, Vogelsang P, Torsvik A, Kvestad KA, Olausson S, Norberg B, Schulz RW, Bogerd J, Santi N, Edvardsen RB, Wargelius A. 2022. Full production cycle performance of gene-edited, sterile Atlantic salmon: growth, smoltification, welfare indicators and fillet composition. Aquaculture 560. [Google Scholar]
- Klug A. 2010. The discovery of zinc fingers and their applications in gene regulation and genome manipulation. Annu Rev Biochem 79: 213–231. [CrossRef] [PubMed] [Google Scholar]
- Kovach RP, Leary RF, Bell DA, Painter S, Lodmell A, Whiteley AR. 2022. Genetic variation in westslope cutthroat trout reveals that widespread genetic rescue is warranted. Can J Fish Aquat Sci 79: 936–946. [CrossRef] [Google Scholar]
- Kozfkay CC, Campbell MR, Heindel JA, Baker DJ, Kline P, Powell MS, Flagg T. 2008. A genetic evaluation of relatedness for broodstock management of captive, endangered Snake River sockeye salmon, Oncorhynchus nerka. Conserv Genet 9: 1421–1430. [CrossRef] [Google Scholar]
- Kriaridou C, Tsairidou S, Fraslin C, Gorjanc G, Looseley ME, Johnston IA, Houston RD, Robledo D. 2023. Evaluation of low-density SNP panels and imputation for cost-effective genomic selection in four aquaculture species. Front Genet 14: 1194266. [CrossRef] [PubMed] [Google Scholar]
- Lacy RC. 2012. Extending pedigree analysis for uncertain parentage and diverse breeding systems. J Hered 103: 197–205. [CrossRef] [PubMed] [Google Scholar]
- Langille BL, Kess T, Brachmann M, Nugent CM, Messmer A, Duffy SJ, Holborn MK, Van Wyngaarden M, Knutsen TM, Kent M, Boyce D, Gregory RS, Gauthier J, Fairchild EA, Pietrak M, Eddy S, de Leaniz CG, Consuegra S, Whittaker B, Bentzen P, Bradbury IR. 2023. Fine-scale environmentally associated spatial structure of lumpfish (Cyclopterus lumpus) across the Northwest Atlantic. Evol Appl 16: 1619–1636. [CrossRef] [PubMed] [Google Scholar]
- Lapègue S, Harrang E, Heurtebise S, Flahauw E, Donnadieu C, Gayral P, Ballenghien M, Genestout L, Barbotte L, Mahla R, Haffray P, Klopp C. 2014. Development of SNP-genotyping arrays in two shellfish species. Mol Ecol Resour 14: 820–830. [CrossRef] [PubMed] [Google Scholar]
- Lapègue S, Reisser C, Harrang E, Heurtebise S, Bierne N. 2023. Genetic parallelism between European flat oyster populations at the edge of their natural range. Evol Appl 16: 393–407. [CrossRef] [PubMed] [Google Scholar]
- Layton KKS, Snelgrove PVR, Dempson JB, Kess T, Lehnert SJ, Bentzen P, Duffy SJ, Messmer AM, Stanley RRE, DiBacco C, Salisbury SJ, Ruzzante DE, Nugent CM, Ferguson MM, Leong JS, Koop BF, Bradbury IR. 2021. Genomic evidence of past and future climate-linked loss in a migratory Arctic fish. Nat Clim Change 11: 158–165. [CrossRef] [Google Scholar]
- Lehnert SJ, Bradbury IR, April J, Wringe BF, Van Wyngaarden M, Bentzen P. 2023. Pre-COSEWIC review of anadromous Atlantic salmon (Salmo salar) in Canada, Part 1: Designatable Units. DFO Can. Sci. Advis. Sec. Res. Doc. 2023/026. iv + 156 p. [Google Scholar]
- Lehnert SJ, Bradbury IR, Wringe BF, Van Wyngaarden M, Bentzen P. 2023. Multifaceted framework for defining conservation units: an example from Atlantic salmon (Salmo salar) in Canada. Evol Appl 16: 1568–1585. [CrossRef] [PubMed] [Google Scholar]
- Lehnert SJ, Kess T, Bentzen P, Kent MP, Lien S, Gilbey J, Clément M, Jeffery NW, Waples RS, Bradbury IR. 2019. Genomic signatures and correlates of widespread population declines in salmon. Nat Commun 10: 2996. [CrossRef] [PubMed] [Google Scholar]
- Lew RM, Finger AJ, Baerwald MR, Goodbla A, May B, Meek MH. 2015. Using next-generation sequencing to assist a conservation hatchery: a single-nucleotide polymorphism panel for the genetic management of endangered delta smelt. T Am Fish Soc 144: 767–779. [CrossRef] [Google Scholar]
- Lhorente JP, Araneda M, Neira R, Yáñez JM. 2019. Advances in genetic improvement for salmon and trout aquaculture: the Chilean situation and prospects. Rev Aquacult 11: 340–353. [CrossRef] [Google Scholar]
- Li XC, Bai YT, Dong Z, Xu CX, Liu SK, Yu H, Kong LF, Li Q. 2023. Chromosome-level genome assembly of the European flat oyster (Ostrea edulis) provides insights into its evolution and adaptation. Comp Biochem Phys D 45: 101045. [Google Scholar]
- Liu K, Feng XY, Ma HJ, Xie N. 2021. Development and characterisation of 13 microsatellite markers of Chanodichthys mongolicus (Cypriniformes: Cyprinidae) by RAD-s eq. J Appl Ichthyol 37: 975–979. [CrossRef] [Google Scholar]
- Liu L, Ang KP, Elliott JAK, Kent MP, Lien S, MacDonald D, Boulding EG. 2017. A genome scan for selection signatures comparing farmed Atlantic salmon with two wild populations: testing colocalisation among outlier markers, candidate genes, and quantitative trait loci for production traits. Evol Appl 10: 276–296. [CrossRef] [MathSciNet] [PubMed] [Google Scholar]
- Liu SX, Palti Y, Gao GT, Rexroad CE. 2016. Development and validation of a SNP panel for parentage assignment in rainbow trout. Aquaculture 452: 178–182. [CrossRef] [Google Scholar]
- Lou RN, Jacobs A, Wilder AP, Therkildsen NO. 2021. A beginner's guide to low-coverage whole genome sequencing for population genomics. Mol Ecol 30: 5966–5993. [CrossRef] [PubMed] [Google Scholar]
- Marchini J, Howie B. 2010. Genotype imputation for genome-wide association studies. Nat Rev Genet 11: 499–511. [CrossRef] [PubMed] [Google Scholar]
- Marcy-Quay B, Wilson CC, Osborne CA, Marsden JE. 2023. Optimisation of an amplicon sequencing-based microsatellite panel and protocol for stock identification and kinship inference of lake trout (Salvelinus namaycush). Ecol Evol 13: e10020. [CrossRef] [PubMed] [Google Scholar]
- Marshall IR, Brauer CJ, Wedderburn SD, Whiterod NS, Hammer MP, Barnes TC, Attard CRM, Möller LM, Beheregaray LB. 2022. Longitudinal monitoring of neutral and adaptive genomic diversity in a reintroduction. Conserv Biol 36: e13889. [CrossRef] [PubMed] [Google Scholar]
- McKenzie JA, Paim U. 1969. Variations in the plasma proteins of Atlantic salmon (Salmo salar L.). Can J Zool 47: 759–761. [CrossRef] [PubMed] [Google Scholar]
- Mendes B, Sampaio T, Antunes MA, Magalhaes H, Silva FCE, Borges C, Simoes F, Usié A, Almeida MH, Ramos AM. 2022. Kinship analysis and pedigree reconstruction of a natural regenerated cork oak (Quercus suber) population. Forests 13. [Google Scholar]
- Meuwissen THE, Hayes BJ, Goddard ME. 2001. Prediction of total genetic value using genome-wide dense marker maps. Genetics 157: 1819–1829. [CrossRef] [PubMed] [Google Scholar]
- Meuwissen THE, Sonesson AK. 2004. Genotype-assisted optimum contribution selection to maximise selection response over a specified time period. Genet Res 84: 109–116. [CrossRef] [Google Scholar]
- Miller J, McLachlan AD, Klug A. 1985. Repetitive zinc-binding domains in the protein transcription factor IIIA from Xenopus oocytes. Embo J 4: 1609–1614. [CrossRef] [PubMed] [Google Scholar]
- Moen T, Baranski M, Sonesson AK, Kjoglum S. 2009. Confirmation and fine-mapping of a major QTL for resistance to infectious pancreatic necrosis in Atlantic salmon (Salmo salar): population-level associations between markers and trait. BMC Genomics 10: 368. [CrossRef] [PubMed] [Google Scholar]
- Moghadam HK, Poissant J, Fotherby H, Haidle L, Ferguson MM, Danzmann RG. 2007. Quantitative trait loci for body weight, condition factor and age at sexual maturation in Arctic charr (Salvelinus alpinus): comparative analysis with rainbow trout (Oncorhynchus mykiss) and Atlantic salmon (Salmo salar). Mol Genet Genomics 277: 647–661. [CrossRef] [PubMed] [Google Scholar]
- Moller D. 1970. Transferrin polymorphism in Atlantic salmon (Salmo salar). J Fish Res Board Can 27: 1617–1625. [CrossRef] [Google Scholar]
- Monk J, Boyce DL, Ang KP, George S, Tucker D, Jeannot K, Fry J, Gianasi B, Hickey S, O'Brien N. 2016. Cleaner fish research and production in Newfoundland. Bull Aquac Assoc Can 2016-2: 39–45. [Google Scholar]
- Morvezen R, Boudry P, Laroche J, Charrier G. 2016. Stock enhancement or sea ranching? Insights from monitoring the genetic diversity, relatedness and effective population size in a seeded great scallop population (Pecten maximus). Heredity 117: 142–148. [CrossRef] [PubMed] [Google Scholar]
- Murray K. 2005. Population genetic assessment of the endangered Atlantic whitefish, Coregonus huntsmani, and the lake whitefish, C. clupeaformis, in Atlantic Canada. MSc. thesis, Dept. of Biology, Dalhousie University, Halifax, NS. [Google Scholar]
- Narum SR, Di Genova A, Micheletti SJ, Maass A. 2018. Genomic variation underlying complex life-history traits revealed by genome sequencing in Chinook salmon. P Roy Soc B-Biol Sci 285. [Google Scholar]
- Neira R, Díaz NF, Gall GAE, Gallardo JA, Lhorente JP, Manterola R. 2006. Genetic improvement in Coho salmon (Oncorhynchus kisutch). I: Selection response and inbreeding depression on harvest weight. Aquaculture 257: 9–17. [CrossRef] [Google Scholar]
- Nguyen NH. 2016. Genetic improvement for important farmed aquaculture species with a reference to carp, tilapia and prawns in Asia: achievements, lessons and challenges. Fish Fish 17: 483–506. [CrossRef] [Google Scholar]
- Nielsen ES, Henriques R, Beger M, von der Heyden S. 2021. Distinct interspecific and intraspecific vulnerability of coastal species to global change. Global Change Biol 27: 3415–3431. [CrossRef] [PubMed] [Google Scholar]
- Norris AT, Bradley DG, Cunningham EP. 2000. Parentage and relatedness determination in farmed Atlantic salmon (Salmo salar) using microsatellite markers. Aquaculture 182: 73–83. [CrossRef] [Google Scholar]
- Nugent CM, Kess T, Brachmann MK, Langille BL, Duffy SJ, Lehnert SJ, Wringe BF, Bentzen P, Bradbury IR. 2023. Whole-genome sequencing reveals fine-scale environment-associated divergence near the range limits of a temperate reef fish. Mol Ecol 32: 4742–4762. [CrossRef] [PubMed] [Google Scholar]
- Nuñez JK, Chen J, Pommier GC, Cogan JZ, Replogle JM, Adriaens C, Ramadoss GN, Shi QM, Hung KL, Samelson AJ, Pogson AN, Kim JYS, Chung A, Leonetti MD, Chang HY, Kampmann M, Bernstein BE, Hovestadt V, Gilbert LA, Weissman JS. 2021. Genome-wide programmable transcriptional memory by CRISPR-based epigenome editing. Cell 184: 2503–2519. [CrossRef] [PubMed] [Google Scholar]
- O'Reilly P. 2006. Towards the identification of Conservation Units in Atlantic salmon from Eastern Canada. DFO Can. Sci. Advis. Sec. Res. Doc. 2006/012. [Google Scholar]
- O'Reilly P, Harvie CJ. 2010. Conservation of genetic variation in the Inner Bay of Fundy Atlantic salmon conservation breeding and rearing program. DFO Can. Sci. Advis. Sec. Res. Doc. 2009/095. [Google Scholar]
- Palaiokostas C, Bekaert M, Davie A, Cowan ME, Oral M, Taggart JB, Gharbi K, McAndrew BJ, Penman DJ, Migaud H. 2013. Mapping the sex determination locus in the Atlantic halibut (Hippoglossus hippoglossus) using RAD sequencing. BMC Genomics 14: 566. [CrossRef] [PubMed] [Google Scholar]
- Payne RH. 1974. Transferrin variation in North American populations of the Atlantic salmon, Salmo salar. J Fish Res Board Can 31: 1037–1041. [CrossRef] [Google Scholar]
- Peace CP. 2017. DNA-informed breeding of rosaceous crops: promises, progress and prospects. Hortic Res-England 4: 17006. [CrossRef] [Google Scholar]
- Peñaloza C, Barria A, Papadopoulou A, Hooper C, Preston J, Green M, Helmer L, Kean-Hammerson J, Nascimento-Schulze JC, Minardi D, Gundappa MK, Macqueen DJ, Hamilton J, Houston RD, Bean TP. 2022. Genome-wide association and genomic prediction of growth traits in the European flat oyster (Ostrea edulis). Front Genet 13: 926638. [CrossRef] [PubMed] [Google Scholar]
- Phelps MP, Seeb LW, Seeb JE. 2020. Transforming ecology and conservation biology through genome editing. Conserv Biol 34: 54–65. [CrossRef] [PubMed] [Google Scholar]
- Pouvreau S, Lapègue S, Arzul I, Boudry P. 2023. Fifty years of research to counter the decline of the European flat oyster (Ostrea edulis): a review of French achievements and prospects for the restoration of remaining beds and revival of aquaculture production. Aquat Living Resour 36. [Google Scholar]
- Prado P, Fernández M, Cordero D, Saavedra C, Carella F, Alcaraz C, Gairin I . 2022. Molecular identification, life cycle characterisation, and hatchery seed production of dwarf oysters from the Ebro Delta (Spain). Aquat Living Resour 35. [Google Scholar]
- Purcell CM, Seetharam AS, Snodgrass O, Ortega-García S, Hyde JR, Severin AJ. 2018. Insights into teleost sex determination from the Seriola dorsalis genome assembly. BMC Genomics 19: 31. [CrossRef] [PubMed] [Google Scholar]
- Qin W, Brown JL. 2006. Consumer opinions about genetically engineered salmon and information effect on opinions: a qualitative approach. Science Communication 28: 243–272. [CrossRef] [Google Scholar]
- Quinton CD, McMillan I, Glebe BD. 2005. Development of an Atlantic salmon (Salmo salar) genetic improvement program: genetic parameters of harvest body weight and carcass quality traits estimated with animal models. Aquaculture 247: 211–217. [CrossRef] [Google Scholar]
- Reid DP, Szanto A, Glebe B, Danzmann RG, Ferguson MM. 2005. QTL for body weight and condition factor in Atlantic salmon (Salmo salar): comparative analysis with rainbow trout (Oncorhynchus mykiss) and Arctic charr (Salvelinus alpinus). Heredity 94: 166–172. [CrossRef] [PubMed] [Google Scholar]
- Rexroad C, Vallet J, Matukumalli LK, Reecy J, Bickhart D, Blackburn H, Boggess M, Cheng H, Clutter A, Cockett N, Ernst C, Fulton JE, Liu J, Lunney J, Neibergs H, Purcell C, Smith TPL, Sonstegard T, Taylor J, Telugu B, Van Eenennaam A, Van Tassell CP, Wells K, Martin A, Murdoch B, Sayre B, Keel B, Schmidt C, Hostetler C, Seabury C, Tuggle C, Elsik C, Gill C, Ciobanu D, Bailey D, Hamernik D, Grings E, Connor E, Rohrer G, Plastow G, Rosa G, Zhou HJ, Koltes J, Decker J, Weller J, Woodward-Greene J, Steibel J, Long J, Lee K, Kuehn L, Worku M, Salem M, McCue M, Serao N, Riggs P, Sponenberg P, Schnabel R, Brooks S, Fernando S, McKay S, Schmitz-Esser S, White S, Lamont S, Kurt T, Palti Y, AAG Community. 2019. Genome to phenome: improving animal health, production, and well-being − a new USDA blueprint for animal genome research 2018–2027. Front Genet 10: 327. [CrossRef] [PubMed] [Google Scholar]
- Robinson NA, Robledo D, Sveen L, Daniels RR, Krasnov A, Coates A, Jin YH, Barrett LT, Lillehammer M, Kettunen AH, Phillips BL, Dempster T, Doeschl-Wilson A, Samsing F, Difford G, Salisbury S, Gjerde B, Haugen JE, Burgerhout E, Dagnachew BS, Kurian D, Fast MD, Rye M, Salazar M, Bron JE, Monaghan SJ, Jacq C, Birkett M, Browman HI, Skiftesvik AB, Fields DM, Selander E, Bui S, Sonesson A, Skugor S, Ostbye TKK, Houston RD. 2023. Applying genetic technologies to combat infectious diseases in aquaculture. Rev Aquacult 15: 491–535. [CrossRef] [PubMed] [Google Scholar]
- Rollinson N, Keith DM, Houde ALS, Debes PV, McBride MC, Hutchings JA. 2014. Risk assessment of inbreeding and outbreeding depression in a captive-breeding program. Conserv Biol 28: 529–540. [Google Scholar]
- Roy S, Kumar V, Behera BK, Parhi J, Mohapatra S, Chakraborty T, Das BK. 2022. CRISPR/Cas genome editing − can it become a game changer in future fisheries sector? Front Mar Sci 9. [Google Scholar]
- Russello MA, Amato G. 2004. Ex situ population management in the absence of pedigree information. Mol Ecol 13: 2829–2840. [CrossRef] [PubMed] [Google Scholar]
- Ryman N, Laikre L. 1991. Effects of supportive breeding on the genetically effective population size. Conserv Biol 5: 325–329. [CrossRef] [Google Scholar]
- Saillant E, Adams N, Lemus JT, Franks JS, Zohar Y, Stubblefield J, Manley C. 2021. First data on aquaculture of the tripletail, Lobotes surinamensis, a promising candidate species for US marine aquaculture. J World Aquacult Soc 52: 582–594. [CrossRef] [Google Scholar]
- Sandoval-Castillo J, Beheregaray LB, Wellenreuther M. 2022. Genomic prediction of growth in a commercially, recreationally, and culturally important marine resource, the Australian snapper (Chrysophrys auratus). G3-Genes Genom Genet 12. [Google Scholar]
- Schaid DJ, Chen WN, Larson NB. 2018. From genome-wide associations to candidate causal variants by statistical fine-mapping. Nat Rev Genet 19: 491–504. [CrossRef] [PubMed] [Google Scholar]
- Schmidt E, Stuart K, Hyde J, Purcell C, Drawbridge M. 2021. Spawning dynamics and egg production characteristics of captive Seriola dorsalis assessed using parentage analyses. Aquac Res 52: 4050–4063. [CrossRef] [Google Scholar]
- Segelbacher G, Bosse M, Burger P, Galbusera P, Godoy JA, Helsen P, Hvilsom C, Iacolina L, Kahric A, Manfrin C, Nonic M, Thizy D, Tsvetkov I, Velickovic N, Vila C, Wisely SM, Buzan E. 2022. New developments in the field of genomic technologies and their relevance to conservation management. Conserv Genet 23: 217–242. [CrossRef] [Google Scholar]
- Serrao NR, Reid SM, Wilson CC. 2018. Conservation genetics of redside dace (Clinostomus elongatus): phylogeography and contemporary spatial structure. Conserv Genet 19: 409–424. [CrossRef] [Google Scholar]
- Simora RMC, Xing D, Bangs XR, Wang WW, Ma XL, Su BF, Khan MGQ, Qin ZK, Lu CY, Alston V, Hettiarachchi D, Johnson A, Li SJ, Coogan M, Gurbatow J, Terhune JS, Wang X, Dunham RA. 2020. CRISPR/Cas9-mediated knock-in of alligator cathelicidin gene in a non-coding region of channel catfish genome. Sci Rep-Uk 10: 22271. [CrossRef] [Google Scholar]
- Sinclair-Waters M, Odegård J, Korsvoll SA, Moen T, Lien S, Primmer CR, Barson NJ. 2020. Beyond large-effect loci: large-scale GWAS reveals a mixed large-effect and polygenic architecture for age at maturity of Atlantic salmon. Genet Sel Evol 52: 9. [CrossRef] [PubMed] [Google Scholar]
- Sirisha M, Sree Ramulu K, Pavan Kumar K, Ramya A, Shiva P, Krishna P, Rushinadha R. 2018. Identification of fish species using DNA barcode from Visakhapatnam, east coast of India. Pharma Innovation 7: 573–579. [Google Scholar]
- Song HL, Hu HX. 2022. Strategies to improve the accuracy and reduce costs of genomic prediction in aquaculture species. Evol Appl 15: 578–590. [CrossRef] [PubMed] [Google Scholar]
- Steele CA, Anderson EC, Ackerman MW, Hess MA, Campbell NR, Narum SR, Campbell MR. 2013. A validation of parentage-based tagging using hatchery steelhead in the Snake River basin. Can J Fish Aquat Sci 70: 1046–1054. [CrossRef] [Google Scholar]
- Stone NM, Kelly AM, Roy LA. 2016. A fish of weedy waters: golden shiner biology and culture. J World Aquacult Soc 47: 152–200. [CrossRef] [Google Scholar]
- Supple MA, Shapiro B. 2018. Conservation of biodiversity in the genomics era. Genome Biol 19: 131. [CrossRef] [PubMed] [Google Scholar]
- Szarmach SJ, Brelsford A, Witt CC, Toews DPL. 2021. Comparing divergence landscapes from reduced-representation and whole genome resequencing in the yellow-rumped warbler (Setophaga coronata) species complex. Mol Ecol 30: 5994–6005. [CrossRef] [PubMed] [Google Scholar]
- Taggart JB, Ferguson A. 1990. Hypervariable minisatellite DNA single locus probes for the Atlantic salmon, Salmo salar L. J Fish Biol 37: 991–993. [CrossRef] [Google Scholar]
- Therkildsen NO, Wilder AP, Conover DO, Munch SB, Baumann H, Palumbi SR. 2019. Contrasting genomic shifts underlie parallel phenotypic evolution in response to fishing. Science 365: 487–490. [CrossRef] [PubMed] [Google Scholar]
- Tringali MD, Bert TM. 1998. Risk to genetic effective population size should be an important consideration in fish stock-enhancement programs. B Mar Sci 62: 641–659. [Google Scholar]
- Tsai HY, Hamilton A, Tinch AE, Guy DR, Gharbi K, Stear MJ, Matika O, Bishop SC, Houston RD. 2015. Genome wide association and genomic prediction for growth traits in juvenile farmed Atlantic salmon using a high density SNP array. BMC Genomics 16: 969. [CrossRef] [PubMed] [Google Scholar]
- Tsai HY, Matika O, Edwards SM, Antolín-Sánchez R, Hamilton A, Guy DR, Tinch AE, Gharbi K, Stear MJ, Taggart JB, Bron JE, Hickey JM, Houston RD. 2017. Genotype imputation to improve the cost-efficiency of genomic selection in farmed Atlantic salmon. G3-Genes Genom Genet 7: 1377–1383. [Google Scholar]
- Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD. 2010. Genome editing with engineered zinc finger nucleases. Nat Rev Genet 11: 636–646. [CrossRef] [PubMed] [Google Scholar]
- Valenza-Troubat N, Davy M, Storey R, Hilario E, Ritchie P, Wellenreuther M. 2022. Differential expression analyses reveal extensive transcriptional plasticity induced by temperature in New Zealand silver trevally (Pseudocaranx georgianus). Evol Appl 15: 237–248. [CrossRef] [PubMed] [Google Scholar]
- Valenza-Troubat N, Montanari S, Ritchie P, Wellenreuther M. 2022. Unravelling the complex genetic basis of growth in New Zealand silver trevally (Pseudocaranx georgianus). G3-Genes Genom Genet 12. [Google Scholar]
- Vallejo RL, Leeds TD, Gao GT, Parsons JE, Martin KE, Evenhuis JP, Fragomeni BO, Wiens GD, Palti Y. 2017. Genomic selection models double the accuracy of predicted breeding values for bacterial cold water disease resistance compared to a traditional pedigree-based model in rainbow trout aquaculture. Genet Sel Evol 49. [PubMed] [Google Scholar]
- Vallejo RL, Silva RMO, Evenhuis JP, Gao GT, Liu SX, Parsons JE, Martin KE, Wiens GD, Lourenco DAL, Leeds TD, Palti Y. 2018. Accurate genomic predictions for BCWD resistance in rainbow trout are achieved using low-density SNP panels: evidence that long-range LD is a major contributing factor. J Anim Breed Genet 135: 263–274. [CrossRef] [Google Scholar]
- Vandeputte M, Haffray P. 2014. Parentage assignment with genomic markers: a major advance for understanding and exploiting genetic variation of quantitative traits in farmed aquatic animals. Front Genet 5: 432. [CrossRef] [PubMed] [Google Scholar]
- Vehviläinen H, Kause A, Koskinen H, Paananen T. 2010. Genetic architecture of rainbow trout survival from egg to adult. Genet Res 92: 1–11. [CrossRef] [PubMed] [Google Scholar]
- Vehviläinen H, Kause A, Kuukka-Anttila H, Koskinen H, Paananen T. 2012. Untangling the positive genetic correlation between rainbow trout growth and survival. Evol Appl 5: 732–745. [CrossRef] [PubMed] [Google Scholar]
- Vehviläinen H, Kause A, Quinton C, Koskinen H, Paananen T. 2008. Survival of the currently fittest: genetics of rainbow trout survival across time and space. Genetics 180: 507–516. [CrossRef] [PubMed] [Google Scholar]
- Vera M, Pardo BG, Cao A, Vilas R, Fernandez C, Blanco A, Gutierrez AP, Bean TP, Houston RD, Villalba A, Martinez P. 2019. Signatures of selection for bonamiosis resistance in European flat oyster (Ostrea edulis): new genomic tools for breeding programs and management of natural resources. Evol Appl 12: 1781–1796. [CrossRef] [PubMed] [Google Scholar]
- Verbyla KL, Kube PD, Evans BS. 2022. Commercial implementation of genomic selection in Tasmanian Atlantic salmon: scheme evolution and validation. Evol Appl 15: 631–644. [CrossRef] [PubMed] [Google Scholar]
- Vu SV, Knibb W, Gondro C, Subramanian S, Nguyen NTH, Alam M, Dove M, Gilmour AR, Vu IV, Bhyan S, Tearle R, Khuong L, Le TS, O'Connor W. 2021. Genomic prediction for whole weight, body shape, meat yield, and color traits in the Portuguese oyster Crassostrea angulata. Front Genet 12: 661276. [CrossRef] [PubMed] [Google Scholar]
- Wang JY, Chen L, Li BJ, Xu J, Feng JX, Dong CJ, Zhou T, Xu P. 2021. Performance of genome prediction for morphological and growth-related traits in Yellow River carp. Aquaculture 536. [Google Scholar]
- Wang QC, Yu Y, Yuan JB, Zhang XJ, Huang H, Li FH, Xiang JH. 2017. Effects of marker density and population structure on the genomic prediction accuracy for growth trait in Pacific white shrimp Litopenaeus vannamei. BMC Genet 18: 45. [CrossRef] [PubMed] [Google Scholar]
- Waples RS, Ford MJ, Nichols K, Kardos M, Myers J, Thompson TQ, Anderson EC, Koch IJ, McKinney G, Miller MR, Naish K, Narum SR, O'Malley KG, Pearse DE, Pess GR, Quinn TP, Seamons TR, Spidle A, Warheit K, Willis SC. 2022. Implications of large-effect loci for conservation: a review and case study with Pacific salmon. J Hered 113: 121–144. [CrossRef] [PubMed] [Google Scholar]
- Waples RS, Naish KA, Primmer CR. 2020. Conservation and management of salmon in the age of genomics. Annu Rev Anim Biosci 8: 117–143. [CrossRef] [PubMed] [Google Scholar]
- Wargelius A, Leininger S, Skaftnesmo KO, Kleppe L, Andersson E, Taranger GL, Schulz RW, Edvardsen RB. 2016. dnd knockout ablates germ cells and demonstrates germ cell independent sex differentiation in Atlantic salmon. Sci Rep-Uk 6: 21284. [CrossRef] [Google Scholar]
- Waters CD, Clemento A, Aykanat T, Garza JC, Naish KA, Narum S, Primmer CR. 2021. Heterogeneous genetic basis of age at maturity in salmonid fishes. Mol Ecol 30: 1435–1456. [CrossRef] [PubMed] [Google Scholar]
- Waters CD, Hard JJ, Brieuc MSO, Fast DE, Warheit KI, Knudsen CM, Bosch WJ, Naish KA. 2018. Genomewide association analyses of fitness traits in captive-reared Chinook salmon: applications in evaluating conservation strategies. Evol Appl 11: 853–868. [CrossRef] [PubMed] [Google Scholar]
- Waters CD, Hard JJ, Brieuc MSO, Fast DE, Warheit KI, Waples RS, Knudsen CM, Bosch WJ, Naish KA. 2015. Effectiveness of managed gene flow in reducing genetic divergence associated with captive breeding. Evol Appl 8: 956–971. [CrossRef] [PubMed] [Google Scholar]
- Waters CD, Hard JJ, Brieuc MSO, Fast DE, Warheit KI, Waples RS, Knudsen CM, Bosch WJ, Naish KA. 2016. What can genomics tell us about the success of enhancement programs in anadromous Chinook salmon? A comparative analysis across four generations. bioRxiv, 087973. [Google Scholar]
- Wellmann R, Bennewitz J, Meuwissen THE. 2014. A unified approach to characterise and conserve adaptive and neutral genetic diversity in subdivided populations. Genet Res 96: e16. [CrossRef] [PubMed] [Google Scholar]
- Wells ZRR, Bernos TA, Yates MC, Fraser DJ. 2019. Genetic rescue insights from population- and family-level hybridisation effects in brook trout. Conserv Genet 20: 851–863. [CrossRef] [Google Scholar]
- Weng ZY, Yang Y, Wang X, Wu LN, Hua SJ, Zhang HF, Meng ZN. 2021. Parentage analysis in giant grouper (Epinephelus lanceolatus) using microsatellite and SNP markers from genotyping-by-sequencing data. Genes-Basel 12(7). [Google Scholar]
- Wenne R. 2023. Single nucleotide polymorphism markers with applications in conservation and exploitation of aquatic natural populations. Animals-Basel 13. [Google Scholar]
- Wong TT, Zohar Y. 2015. Production of reproductively sterile fish: a mini-review of germ cell elimination technologies. Gen Comp Endocr 221: 3–8. [CrossRef] [Google Scholar]
- Wood G, Marzinelli EM, Campbell AH, Steinberg PD, Vergés A, Coleman MA. 2021. Genomic vulnerability of a dominant seaweed points to future-proofing pathways for Australia's underwater forests. Global Change Biol 27: 2200–2212. [CrossRef] [PubMed] [Google Scholar]
- Wringe BF, Devlin RH, Ferguson MM, Moghadam HK, Sakhrani D, Danzmann RG. 2010. Growth-related quantitative trait loci in domestic and wild rainbow trout (Oncorhynchus mykiss). BMC Genet 11. [PubMed] [Google Scholar]
- Xuereb A, Kimber CM, Curtis JMR, Bernatchez L, Fortin MJ. 2018. Putatively adaptive genetic variation in the giant California sea cucumber (Parastichopus californicus) as revealed by environmental association analysis of restriction-site associated DNA sequencing data. Mol Ecol 27: 5035–5048. [CrossRef] [PubMed] [Google Scholar]
- Yáñez JM, Xu P, Carvalheiro R, Hayes B. 2022. Genomics applied to livestock and aquaculture breeding. Evol Appl 15: 517–522. [CrossRef] [PubMed] [Google Scholar]
- Yoshida GM, Carvalheiro R, Lhorente JP, Correa K, Figueroa R, Houston RD, Yáñez JM. 2018. Accuracy of genotype imputation and genomic predictions in a two-generation farmed Atlantic salmon population using high-density and low-density SNP panels. Aquaculture 491: 147–154. [CrossRef] [Google Scholar]
- Yoshida GM, Lhorente JP, Correa K, Soto J, Salas D, Yáñez JM. 2019. Genome-wide association study and cost-efficient genomic predictions for growth and fillet yield in Nile tilapia (Oreochromis niloticus). G3-Genes Genom Genet 9: 2597–2607. [CrossRef] [Google Scholar]
- Zeng QF, Zhao BJ, Wang H, Wang MQ, Teng MX, Hu JJ, Bao ZM, Wang YF. 2022. Aquaculture molecular breeding platform (AMBP): a comprehensive web server for genotype imputation and genetic analysis in aquaculture. Nucleic Acids Res 50(W1), W66– W74. [CrossRef] [PubMed] [Google Scholar]
- Zu Ermgassen PSE, Strand A, Bakker N, Blanco A, Bonacic K, Boudry P, Brundu G, Cameron TC, Connellan I, da Costa F, Debney A, Fabra M, Frankic A, Gamble C, Gray MW, Helmer L, Holbrook Z, Hugh-Jones T, Kamermans P, Magnesen T, Nielsen P, Preston J, Ranger CJ, Saurel C, Smyth D, Stechele B, Theodorou JA, Colsoul B 2023. Overcoming Ostrea edulis seed production limitations to meet ecosystem restoration demands in the UN decade on restoration. Aquat Living Resour 36. [Google Scholar]
Cite this article as: Purcell CM, Wringe BF, Boudry P, Brieuc MSO, Coulson MW, Kess T, Solberg MF, Vehviläinen H, Calboli FCF. 2025. States of development and application of genetic and genomic tools in aquaculture and conservation programs: A guide for strengthening dialogue among practitioners of aquaculture and genetics. Aquat. Living Resour. 38: 11. https://doi.org/10.1051/alr/2025006
All Tables
States, and state skipping, describing the development and application of genetic and genomic tools in aquaculture and conservation breeding programs, with selected examples under each category as available.
All Figures
|
Fig. 1 Decision trees that aquaculture program managers must consider to guide development of their operations. These decisions range from choices regarding organism selection, research strategies, and considerations of economic and technological limitations for both commercial and restoration-focused breeding programs. Coloured nodes refer to the States of development and application of genetic and genomic tools, and correspond with the colors and States described in Table 1. |
| In the text | |
|
Fig. 2 The six large panels represent the States of development, the application of genetic and genomic tools, and broad research categories associated with each State. Inset are the relative levels (low, medium, or high − depicted by one, two, or three icons, respectively) of resource investment likely required to address research at each State: sampling or phenotyping labour, breeding program infrastructure (e.g., tanks, labour), genetic or genomic labouratory support (e.g., equipment and specialised labour), and bioinformatic support (e.g., computational and bioinformatics labour). While the requirements for each of these categories of resources may vary widely across different programs at each of the States, the relative levels are intended to depict a minimum level of resource investment that new adopters could expect with research or operations conducted at each of these States. |
| In the text | |
|
Fig. 3 Schematic illustration of the utility of an annotated, whole genome in aquaculture or conservation breeding programs. Solid arrows represent directionality of information or activity with colours denoting common pathways. The grey dashed line, representing traditional selection, is included to reinforce that it operates primarily based on phenotypes and that it can occur while genomic tools are being developed. |
| In the text | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.